Overview
- The ancient DNA revolution, pioneered by Svante Pääbo and recognised with the 2022 Nobel Prize, has transformed the study of human population history by enabling direct genomic analysis of individuals who lived thousands of years ago, revealing migration events and admixture patterns invisible to archaeology alone.
- Key discoveries include the identification of three ancestral populations underlying modern European genomes (Western Hunter-Gatherers, Early European Farmers, and Steppe pastoralists), the massive Yamnaya-related expansion that reshaped Bronze Age Europe, and the complex multi-wave peopling of the Americas from a Beringian standstill population.
- Methodological advances such as petrous bone extraction, single-stranded library preparation, and capture enrichment have expanded the ancient DNA record to more than 10,000 published ancient genomes curated in the Allen Ancient DNA Resource, while growing ethical frameworks emphasise Indigenous community consultation and the repatriation of ancestral remains.
Ancient DNA (aDNA) research has fundamentally transformed the study of human population history. Where once scholars relied on the morphology of bones, the distribution of artefacts, and the comparative analysis of living populations to infer past migrations, paleogenomics now provides direct access to the genomes of individuals who lived hundreds or thousands of years ago. The field's modern foundations were laid by Svante Pääbo, whose early work on Egyptian mummy DNA in 1985 and whose laboratory's landmark sequencing of the Neanderthal genome in 2010 demonstrated that ancient genomes could yield information unobtainable by any other means.1, 2 Pääbo's contributions were recognised with the 2022 Nobel Prize in Physiology or Medicine, awarded for his discoveries concerning the genomes of extinct hominins and human evolution.3
Since the Neanderthal genome project, the pace of ancient DNA discovery has accelerated dramatically. Technological innovations in DNA extraction, library preparation, and targeted enrichment have made it possible to recover genome-wide data from skeletal remains preserved in a wide range of climates and depositional contexts. By 2022, more than 10,000 ancient human genomes had been published, curated in the Allen Ancient DNA Resource (AADR), transforming understanding of population movements, admixture events, social organisation, and natural selection across every inhabited continent.24
Origins of the ancient DNA revolution
The recovery of genetic information from ancient biological material was first demonstrated in the 1980s. In 1985, Svante Pääbo reported the molecular cloning of DNA from a 2,400-year-old Egyptian mummy, extracting approximately 3.4 kilobases of human DNA including members of the Alu repetitive element family.1 This work established that DNA could survive in preserved tissues over millennia, though the field spent the next two decades grappling with the pervasive problem of contamination by modern DNA, which could easily overwhelm the tiny quantities of endogenous ancient molecules present in most archaeological specimens.

{kind=link}
The decisive breakthrough came with the application of next-generation sequencing (NGS) technologies in the late 2000s. Unlike earlier PCR-based approaches that amplified specific target sequences and were highly vulnerable to contamination, NGS platforms could sequence all DNA molecules in a library simultaneously, allowing researchers to distinguish authentic ancient sequences from modern contaminants on the basis of characteristic damage patterns, particularly the accumulation of cytosine-to-uracil deamination at fragment termini.5, 6 In 2010, Rasmussen and colleagues published the first complete ancient human genome, sequenced to approximately 20-fold coverage from a 4,000-year-old permafrost-preserved hair sample of a Palaeo-Eskimo individual from Greenland.23 That same year, Green and colleagues in Pääbo's laboratory published the draft Neanderthal genome, sequenced from three bones recovered from Vindija Cave in Croatia, revealing that non-African modern humans carry approximately 1 to 4 percent Neanderthal ancestry — definitive evidence of interbreeding between modern humans and Neanderthals after the out-of-Africa dispersal.2
These achievements established paleogenomics as a rigorous discipline, and the 2022 Nobel Prize in Physiology or Medicine awarded to Pääbo recognised the field's transformation of the study of human evolution and population history.3
Laboratory methods and technical advances
The expansion of ancient DNA research from a handful of exceptional specimens to thousands of individuals per year has depended on a series of methodological innovations that dramatically increased DNA recovery rates and reduced the cost per genome. These advances operate at every stage of the workflow, from the initial sampling of skeletal material to the final computational analysis of sequence data.
The single most important advance in sampling strategy was the discovery that the petrous portion of the temporal bone — the densest bone in the mammalian skeleton, encasing the structures of the inner ear — preserves endogenous DNA at concentrations far exceeding those found in other skeletal elements. In 2015, Pinhasi and colleagues demonstrated that the inner ear portion of the petrous bone (the otic capsule) can yield endogenous DNA fractions up to 65-fold higher than the surrounding cortical bone and up to 177-fold higher than trabecular bone from the same specimen.4 This discovery transformed the practical economics of ancient DNA research: specimens that previously required prohibitively deep sequencing to recover a usable fraction of human DNA could now yield genome-wide data at modest cost.
At the library preparation stage, the development of single-stranded DNA library protocols by Gansauge and Meyer in 2013 represented a substantial improvement over earlier double-stranded methods. Because ancient DNA is heavily fragmented and chemically damaged, with typical fragment lengths of 30 to 80 base pairs, single-stranded library preparation captures a larger fraction of these ultrashort molecules by ligating adapters to individual denatured strands rather than requiring intact double-stranded ends.5 Combined with partial uracil-DNA-glycosylase (UDG) treatment — an enzymatic step that removes uracil residues from the interior of ancient DNA molecules while preserving the terminal damage pattern used for authentication — this approach simultaneously improves sequence quality and retains the molecular signatures needed to verify the ancient origin of the DNA.6
Finally, in-solution capture enrichment has enabled researchers to target specific subsets of the genome, most commonly a panel of approximately 1.24 million single nucleotide polymorphisms (SNPs) that are informative for population genetic analysis. Rather than sequencing the entire genome at low coverage, capture enrichment concentrates sequencing effort on the most informative sites, reducing per-sample costs by an order of magnitude and enabling the large-scale studies of hundreds or thousands of individuals that have characterised the field since 2015.8, 10
The triple ancestry of modern Europeans
{kind=link}
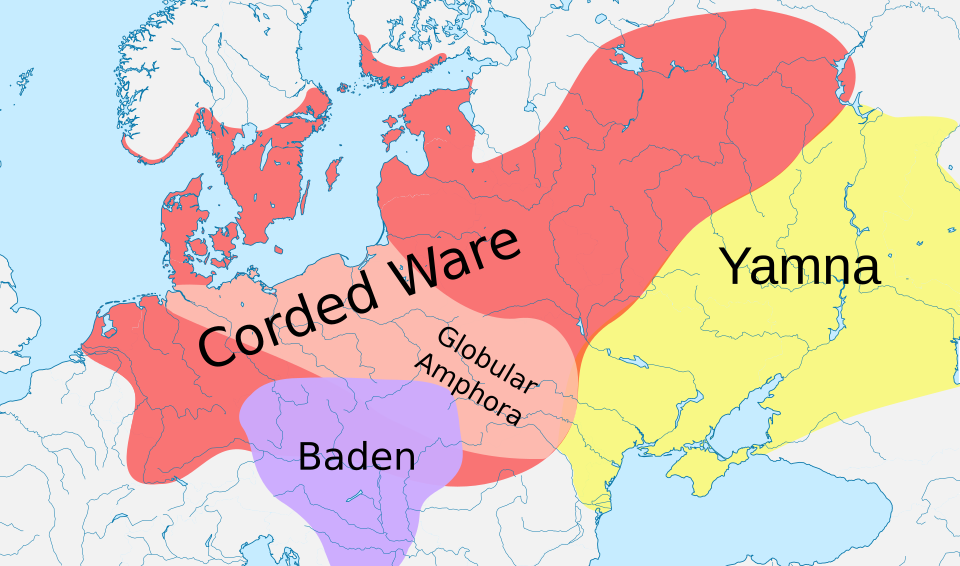
One of the most transformative findings of the ancient DNA era has been the elucidation of the deep population structure underlying modern European genomes. In 2014, Lazaridis and colleagues demonstrated through analysis of ancient genomes from across Europe and western Asia that most present-day Europeans derive their ancestry from at least three highly differentiated source populations: Western Hunter-Gatherers (WHG), who inhabited Europe after the Last Glacial Maximum; Early European Farmers (EEF), who migrated into Europe from the Near East beginning around 7000 BCE and brought the Neolithic agricultural package; and a third component related to Ancient North Eurasians (ANE), which arrived later and is closely connected to the Steppe pastoralist populations.7
This three-component model replaced earlier, simpler narratives that had posited either continuity between Mesolithic hunter-gatherers and modern Europeans or a simple two-way admixture between hunter-gatherers and incoming farmers. The ancient DNA data revealed that the arrival of farming in Europe was accompanied by a substantial demographic replacement: Early European Farmers were genetically distinct from the indigenous hunter-gatherers, deriving the majority of their ancestry from populations originating in Anatolia and the Levant. The transition to farming was not primarily a diffusion of ideas and technologies, but a migration of people who largely replaced the pre-existing hunter-gatherer populations, although some admixture between the two groups occurred over subsequent millennia.7, 21
The third ancestral component — Steppe-related ancestry — was not present in Europe during the Neolithic and only appeared in the archaeological record beginning around 3000 BCE, associated with the Corded Ware and subsequent Bell Beaker cultural complexes. Its identification required the analysis of Bronze Age genomes, which revealed the massive demographic impact of the Yamnaya expansion described below.8, 9
The Yamnaya expansion and the transformation of Bronze Age Europe
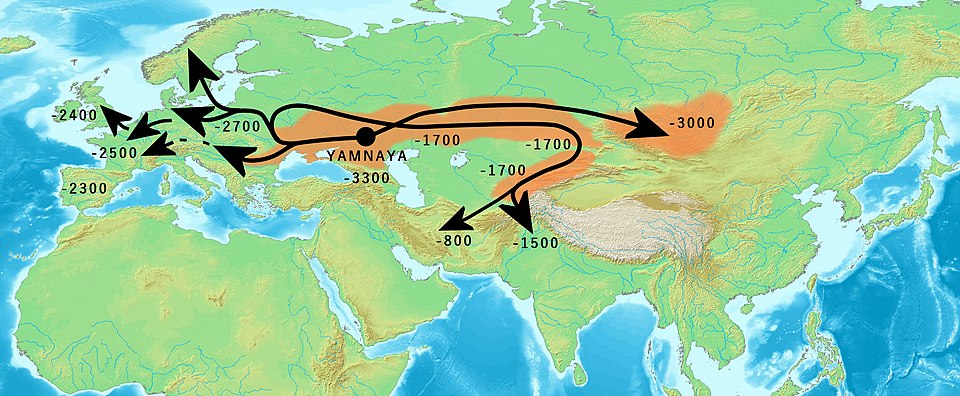
In 2015, two landmark studies published simultaneously in Nature provided genome-wide evidence for a massive migration from the Pontic-Caspian steppe into central and western Europe during the third millennium BCE. Haak and colleagues generated genome-wide data from 69 Europeans spanning the period from 8000 to 3000 BCE, demonstrating that individuals associated with the Late Neolithic Corded Ware culture in Germany derived approximately 75 percent of their ancestry from populations related to the Yamnaya pastoralists of the Pontic-Caspian steppe.8 Independently, Allentoft and colleagues sequenced 101 ancient genomes from across Eurasia and reached concordant conclusions, showing that the Bronze Age was a period of large-scale population migrations and replacements that fundamentally reshaped the demographic structure of both Europe and central Asia.9
{kind=link}
The Yamnaya (from the Russian for "pit grave," referring to their characteristic burial practice) were semi-nomadic pastoralists who herded cattle, sheep, and horses on the grasslands north of the Black and Caspian seas from approximately 3300 to 2600 BCE. Ancient DNA analysis revealed that their genetic profile combined two deeply divergent ancestral components: one related to Eastern European and Caucasus hunter-gatherers, and another with affinities to populations from the Near East and Iran.8, 9 Beginning around 3000 BCE, Yamnaya-related populations expanded rapidly both westward into Europe and eastward across the Eurasian steppe, leaving a genetic signature that persists in virtually all present-day European populations.
The scale of the demographic transformation was most dramatically illustrated in Britain, where Olalde and colleagues demonstrated in 2018 that the arrival of the Bell Beaker complex around 2450 BCE was accompanied by the replacement of approximately 90 percent of Britain's existing gene pool within a few centuries. The incoming Beaker-associated individuals carried high proportions of Steppe-related ancestry, and their genetic profile rapidly came to dominate the island.11 This finding was particularly striking because the Bell Beaker complex had long been debated in archaeology — whether it represented the movement of people or merely the diffusion of objects and ideas. The ancient DNA evidence was unambiguous: at least in Britain, the Beaker phenomenon was accompanied by a near-total population replacement.
The Yamnaya expansion also carried important implications for historical linguistics: both Haak et al. and Allentoft et al. noted that the timing and geography of the Steppe migration were consistent with the hypothesis that the Indo-European language family originated among Steppe pastoralist populations and spread westward through demographic expansion rather than purely cultural transmission.8, 9
Major ancestral components of modern European populations7, 8, 10
| Ancestral component | Origin | Arrival in Europe | Key associated cultures | Contribution to modern Europeans |
|---|---|---|---|---|
| Western Hunter-Gatherers (WHG) | Post-glacial Europe | Indigenous (post-LGM recolonisation) | Mesolithic forager groups | ~10–30% |
| Early European Farmers (EEF) | Anatolia / Near East | ~7000–5000 BCE | LBK, Cardial Ware | ~25–50% |
| Steppe pastoralists (Yamnaya-related) | Pontic-Caspian steppe | ~3000–2500 BCE | Corded Ware, Bell Beaker | ~20–50% |
The peopling of the Americas
Ancient DNA has also substantially revised the understanding of how and when humans first colonised the Americas. Raghavan and colleagues published genome-wide data from both ancient and modern Native American populations in 2015, providing evidence that all Native Americans descended from a single founding population that diverged from East Asian ancestors and experienced a prolonged period of genetic isolation — the so-called Beringian standstill — before entering the Americas.12 This model, originally proposed on the basis of mitochondrial DNA variation, was confirmed and refined by whole-genome analysis: the ancestral Native American population split from East Asian populations roughly 23,000 years ago and remained genetically isolated in Beringia for several thousand years before dispersing southward into the Americas no earlier than approximately 16,000 years ago.12
Subsequent ancient DNA studies revealed unexpected complexity in the initial colonisation. Moreno-Mayar and colleagues sequenced 15 ancient human genomes spanning from Alaska to Patagonia, including six individuals older than 10,000 years, and demonstrated that the initial dispersal was followed by rapid diversification into multiple genetically distinct populations, some of which represent previously unknown groups with no clear modern descendants.13 Their analysis also detected evidence of a cryptic Australasian-related genetic signal in some ancient South American populations, the origin of which remains debated but which may reflect an early migration event distinct from the primary wave that gave rise to most Native American populations.13
The genomic study of the Kennewick Man (also known as the Ancient One), an approximately 8,500-year-old skeleton discovered along the Columbia River in Washington State in 1996, illustrates both the scientific power and the ethical complexity of ancient DNA research in the Americas. After decades of legal dispute between scientists and the Confederated Tribes of the Colville Reservation and other claimant tribes, Rasmussen and colleagues sequenced the Kennewick Man genome in 2015 and demonstrated that the individual was genetically closer to modern Native Americans than to any other population worldwide, supporting the tribes' claims of ancestral affiliation and eventually facilitating repatriation under the Native American Graves Protection and Repatriation Act (NAGPRA).14
The Austronesian and Bantu expansions
Beyond Europe and the Americas, ancient DNA has illuminated two of the most consequential demographic expansions of the Holocene: the Austronesian dispersal across Island Southeast Asia and the Pacific, and the Bantu expansion across sub-Saharan Africa. Both events reshaped the genetic, linguistic, and cultural landscape of vast regions, and both had been debated for decades on the basis of linguistic and archaeological evidence before ancient DNA provided direct resolution.
In Southeast Asia, Lipson and colleagues generated genome-wide ancient DNA from 18 individuals spanning the Neolithic through the Iron Age (approximately 4100 to 1700 years ago) and documented at least two major waves of genetic admixture: the first between local hunter-gatherers and incoming farmers with ancestry tracing to southern China, associated with the initial spread of agriculture, and a second pulse of Chinese-related ancestry arriving during the Bronze Age.15 These findings provided genomic support for the "out of Taiwan" model of Austronesian expansion, in which farming populations originating in southern China and Taiwan expanded southward through the Philippines and Indonesia beginning around 4000 years ago, mixing with but substantially replacing the indigenous hunter-gatherer populations of Island Southeast Asia.15
In Africa, where the preservation of ancient DNA is particularly challenging due to tropical and subtropical climates that accelerate DNA degradation, a series of studies beginning with Skoglund and colleagues in 2017 assembled the first genome-wide ancient DNA data from the continent. Analysis of 16 prehistoric African genomes revealed that the deep population structure of the continent was far more complex than modern genetic diversity alone had suggested. For example, ancient hunter-gatherers from Malawi dating to approximately 8,100 to 2,500 years ago derived roughly two-thirds of their ancestry from a deeply divergent lineage related to the modern southern African San, a lineage that has since been largely displaced across eastern and central Africa by the expansion of Bantu-speaking farming populations over the past 3,000 to 4,000 years.16 Prendergast and colleagues extended this picture in 2019, using ancient DNA from East African pastoral sites to demonstrate that the spread of herding into sub-Saharan Africa occurred through multiple distinct migration events, with incoming pastoralists mixing with local forager populations in stages.17
Bronze Age plague and population turnover
One of the most unexpected contributions of ancient DNA has been the detection of epidemic pathogens in the bones and teeth of prehistoric individuals, revealing episodes of infectious disease that left no trace in the written record. The bacterium Yersinia pestis, the causative agent of plague, has been identified in multiple ancient European populations spanning the Neolithic through the Bronze Age, raising the possibility that pandemic disease contributed to major demographic transitions previously attributed solely to migration or cultural change.
Rascovan and colleagues reported in 2019 the discovery and genome reconstruction of Y. pestis in Neolithic farmers from Sweden dating to approximately 4,900 years ago, representing a lineage basal to all previously known ancient and modern strains of the pathogen. Their analysis suggested that multiple independent lineages of Y. pestis branched and expanded across Eurasia during the late Neolithic, a period corresponding to a well-documented decline in Neolithic farming populations in Europe. The authors proposed that a prehistoric plague pandemic may have contributed to the population collapse that preceded and perhaps facilitated the Yamnaya expansion into a partially depopulated landscape.18
Spyrou and colleagues independently reconstructed Y. pestis genomes from approximately 3,800-year-old Bronze Age individuals from the Samara region of Russia, documenting the emergence of key virulence adaptations — including the acquisition of the ymt gene required for flea-borne transmission — that would later characterise the medieval Black Death. Their findings suggested that the Bronze Age represented a critical period in the evolution of plague, during which the pathogen transitioned from a primarily pneumonic or direct-contact mode of transmission to the flea-borne bubonic form that would cause devastating pandemics in historical times.19
Kinship studies and social organisation
Ancient DNA has also opened a new window onto the social structures of past communities by enabling the direct identification of biological relationships among individuals buried in the same cemetery or household. Traditional archaeological approaches to kinship relied on the spatial arrangement of burials, the distribution of grave goods, and ethnographic analogy; ancient genomics now provides unambiguous identification of parent-child, sibling, and more distant biological relationships, as well as the detection of patterns such as patrilocality (males remaining in their birth community while females move between communities) and matrilocality (the reverse).
A landmark study by Mittnik and colleagues in 2019 combined genome-wide data from 104 individuals buried in farmstead-related cemeteries in the Lech River valley of southern Germany, spanning from the Late Neolithic through the Middle Bronze Age (approximately 2750 to 1300 BCE). The analysis revealed that individual households persisted for multiple generations, with a consistent pattern: the core family members were genetically related through the male line and buried with elaborate grave goods, while genetically unrelated individuals in the same household were buried without significant markers of status.20 Women in the high-status core families were not locally born, as indicated by both their genetic distinctiveness from the local male lineages and strontium isotope ratios in their teeth reflecting non-local geological signatures. The results documented a system of patrilocal residence and female exogamy combined with inherited social inequality that remained stable for approximately 700 years.20
Genome-wide ancient DNA data from the Neolithic through the Bronze Age in Hungary similarly revealed episodes of genetic stability punctuated by dramatic population turnover, with the advent of the Neolithic, Bronze, and Iron Ages each marked by the arrival of genetically distinct populations.21 These findings have demonstrated that kinship analysis through ancient DNA can reconstruct social dynamics — patterns of marriage, inheritance, and social stratification — with a resolution that was previously inaccessible to either archaeology or genetics alone.
Detecting natural selection through time
A unique advantage of the ancient DNA time series is the ability to observe allele frequency changes directly across generations, providing a powerful approach for detecting natural selection that complements methods based on patterns in living populations. Mathieson and colleagues assembled genome-wide data from 230 ancient West Eurasians spanning the period from 6500 to 300 BCE and conducted a systematic genome-wide scan for evidence of selection.10
The analysis detected strong selection at multiple loci, with several findings that overturned previous assumptions. Selection for lighter skin pigmentation in Europe, driven by alleles at the SLC24A5 and SLC45A2 genes, was already largely complete by the Bronze Age, confirming that the light skin characteristic of modern northern Europeans was established relatively recently in evolutionary terms. However, the allele for lactose tolerance in adults, associated with the LCT gene, was still rare in Bronze Age Europeans despite being common in modern northern European populations, indicating that the intense positive selection for this trait occurred primarily during the last 4,000 years — substantially more recently than some models had predicted.9, 10 The study also detected two independent episodes of selection for increased height at different time periods, as well as signals of selection at immune-related genes, likely driven by changing pathogen pressures as populations shifted from hunting and gathering to farming and increasingly dense settlement.10
Ethical considerations and community engagement
The rapid expansion of paleogenomics has raised significant ethical questions about the treatment of ancestral human remains, the rights of descendant communities, and the responsibilities of researchers. In the United States, the Native American Graves Protection and Repatriation Act (NAGPRA), enacted in 1990, provides a legal framework for the repatriation of Native American human remains, funerary objects, and sacred items held by federally funded institutions. However, the intersection of NAGPRA with ancient DNA research has generated considerable tension, as illustrated by the decades-long Kennewick Man dispute, in which scientific interest in studying an exceptionally old skeleton conflicted with tribal claims to ancestral remains.14
In 2018, Bardill and colleagues, writing on behalf of the Summer Internship for Indigenous Peoples in Genomics (SING) Consortium, published a set of recommendations for advancing the ethics of paleogenomics. Their framework emphasised that ancestral remains should be regarded not as archaeological artefacts but as human relatives deserving of respect, and that DNA results from ancient individuals can have significant implications for descendant communities — potentially undermining or complicating claims in treaty, repatriation, territorial, or other legal contexts.22 The authors called for meaningful consultation with descendant communities before research begins, for the inclusion of community representatives in the research process, and for the recognition that communities have the right to decline participation in genetic studies of their ancestors.22
These ethical principles are increasingly reflected in the policies of major ancient DNA laboratories and funding agencies, though implementation remains uneven across different national and institutional contexts. The tension between the scientific imperative to study ancient genomes and the legitimate interests of descendant communities continues to shape the practice of paleogenomics worldwide.
The Allen Ancient DNA Resource and the future of the field
The sheer volume of published ancient genome data has necessitated the creation of centralised, curated databases. The Allen Ancient DNA Resource (AADR), maintained by the Reich laboratory at Harvard University and funded by the Allen Discovery Center, provides the most comprehensive publicly available compilation of ancient human genomes. As of its sixth public release (version 54.1), the AADR contains genome-wide data from more than 10,000 ancient individuals, assayed at more than one million SNPs, alongside comparative data from thousands of present-day individuals. The resource is freely available with version-controlled releases and permanent digital object identifiers for each version, enabling reproducibility across studies.24
The AADR has become an essential infrastructure for the field, enabling meta-analyses that synthesise data from hundreds of independent studies and facilitating the detection of patterns — such as continent-wide migration events, subtle admixture signals, and temporal trends in allele frequencies — that would be invisible in any single dataset. The continued expansion of the resource, as ancient DNA sampling extends into regions that have been underrepresented in the early years of the field, including sub-Saharan Africa, South and Southeast Asia, and Oceania, promises to provide an increasingly global perspective on human population history.16, 17, 24
Looking ahead, ongoing technical improvements in DNA recovery from extremely degraded specimens, the application of imputation algorithms to low-coverage ancient genomes, and the integration of ancient DNA data with isotopic, archaeological, and linguistic evidence continue to expand the scope and resolution of paleogenomic research. The field that Pääbo inaugurated with a single Egyptian mummy now encompasses a global time series of human genetic variation spanning the last 45,000 years, providing an empirical foundation for understanding how the populations and social structures of the modern world came into being.1, 3, 24
References
Genomic evidence for the Pleistocene and recent population history of Native Americans
Emergence and spread of basal lineages of Yersinia pestis during the Neolithic decline
Analysis of 3800-year-old Yersinia pestis genomes suggests Bronze Age origin for bubonic plague