Overview
- Genetic hitchhiking occurs when a selectively neutral allele increases in frequency because it is physically linked on the same chromosome to a positively selected allele undergoing a selective sweep, reducing genetic diversity at linked sites.
- Maynard Smith and Haigh's 1974 model demonstrated that positive selection on one locus can drastically reduce variation at neighbouring loci, and subsequent work distinguished hard sweeps (from new mutations) from soft sweeps (from standing variation), each leaving distinct genomic signatures.
- Detection methods including Tajima's D, Fay and Wu's H, and extended haplotype homozygosity tests have identified hitchhiking signatures around genes such as LCT (lactase persistence) and SLC24A5 (skin pigmentation), confirming that selective sweeps have shaped human and other genomes.
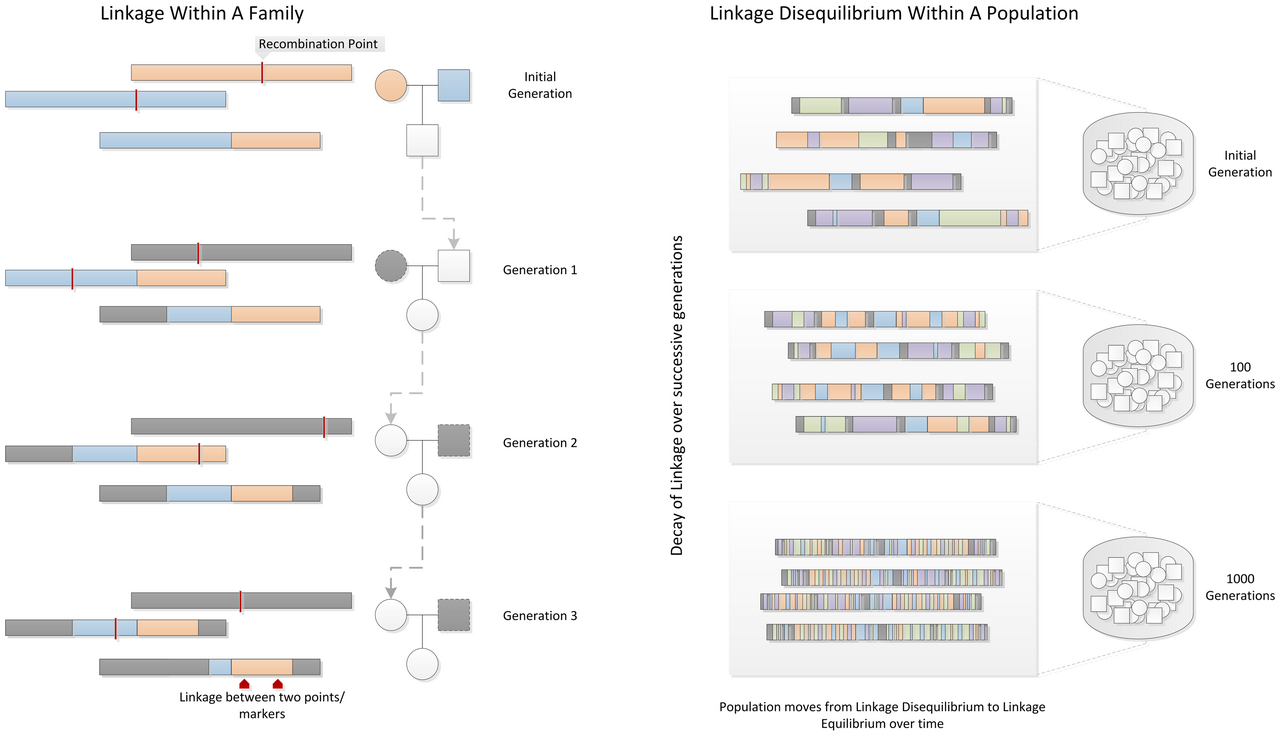
Genetic hitchhiking is the process by which an allele at a selectively neutral locus changes in frequency because it is physically linked to another allele that is under positive natural selection. When a beneficial mutation arises on a chromosome and sweeps to fixation, it carries along all the neutral and nearly neutral variants that happen to reside on the same chromosomal segment, much as passengers on a bus are transported to the bus's destination regardless of their individual intentions. The concept was first formalised by John Maynard Smith and John Haigh in their landmark 1974 paper, which demonstrated that selective sweeps can dramatically reduce genetic variation at linked sites and produce characteristic distortions in the frequency spectrum of neutral polymorphisms.1 Genetic hitchhiking and its inverse, background selection, are now recognised as major forces shaping patterns of nucleotide diversity across genomes, and the detection of hitchhiking signatures has become one of the principal methods for identifying loci that have experienced recent positive selection.1, 11
{kind=link}
The Maynard Smith and Haigh model
In their 1974 paper "The hitch-hiking effect of a favourable gene," Maynard Smith and Haigh considered a simple scenario: a new beneficial mutation arises in a large population and increases in frequency under directional selection until it reaches fixation. They asked what happens to variation at a linked neutral locus during this process. Their analysis showed that as the beneficial allele sweeps through the population, it drags linked neutral variants to high frequency along with it, reducing the effective number of distinct haplotypes in the population and thereby decreasing nucleotide diversity at the linked locus.1
The magnitude of the diversity reduction depends on the recombination rate between the selected and neutral loci. If the two loci are tightly linked (low recombination), the neutral locus is almost completely "swept" along with the beneficial allele, and diversity at the neutral site is reduced nearly to zero. As the recombination distance increases, the probability that a recombination event will decouple the neutral allele from the sweeping beneficial allele also increases, and the reduction in diversity becomes progressively weaker. Maynard Smith and Haigh showed that the expected heterozygosity at a neutral locus a recombinational distance r from the selected site, after a completed sweep of a beneficial allele with selection coefficient s, is approximately H0 × (1 − 2s/(2s + r)), where H0 is the pre-sweep heterozygosity. This means that the hitchhiking effect extends over a chromosomal region whose size is proportional to the ratio s/r: stronger selection and lower recombination both increase the footprint of the sweep.1, 14
The hitchhiking model was initially proposed in part to explain an empirical puzzle: observed levels of genetic variation in Drosophila populations were far lower than predicted by the neutral theory, which expected diversity to scale with population size. Maynard Smith and Haigh suggested that recurrent selective sweeps throughout the genome could periodically purge neutral variation at linked sites, keeping diversity below the neutral equilibrium even in species with very large census population sizes.1, 19
Hard sweeps versus soft sweeps
The classic hitchhiking model describes what is now called a hard sweep: a single new beneficial mutation arises on a single chromosomal background and rises to fixation, dragging that specific haplotype to high frequency and leaving a pronounced valley of reduced diversity centred on the selected site. Hard sweeps produce the most dramatic genomic signatures because all copies of the beneficial allele in the post-sweep population trace back to a single ancestral chromosome.12
Hermisson and Pennings introduced the concept of soft sweeps in 2005 to describe adaptation from standing genetic variation or from recurrent mutation. In a soft sweep, the beneficial allele is already present on multiple chromosomal backgrounds before selection begins, either because it segregated as a neutral or mildly deleterious variant before an environmental change made it advantageous, or because it arose independently by mutation in multiple individuals. When such an allele is driven to fixation by selection, it carries with it multiple distinct haplotypes rather than a single one, producing a less pronounced reduction in diversity at linked neutral sites.12
The distinction between hard and soft sweeps has important implications for the detection of positive selection in genomic data. Hard sweeps leave a characteristic signature of severely reduced diversity, elevated linkage disequilibrium, and a skewed site frequency spectrum in the region surrounding the selected locus. Soft sweeps, by contrast, leave a more subtle footprint: diversity is reduced but not eliminated, and the pattern of haplotype structure is less extreme. Messer and Petrov argued that soft sweeps may be far more common than hard sweeps, particularly in large populations and in species that adapt primarily from standing variation rather than new mutation, which would mean that many instances of positive selection leave only faint or cryptic genomic signatures.13
Genomic signatures of selective sweeps
A completed hard sweep produces several distinctive patterns in genomic data. First, nucleotide diversity is dramatically reduced in the region flanking the selected site, because the sweep has replaced most of the pre-existing haplotypic variation with a single ancestral haplotype. Second, the site frequency spectrum is distorted: there is an excess of rare variants (because new mutations have accumulated since the sweep on the now-homogeneous background) and, in some cases, an excess of high-frequency derived variants (because the sweep carried derived alleles at linked sites to near-fixation). Third, linkage disequilibrium is elevated in the swept region because recombination has not yet had time to break apart the extended haplotype generated by the sweep.1, 2, 4
An ongoing or incomplete sweep produces a different but equally diagnostic pattern. Because the beneficial allele has not yet reached fixation, there is a marked contrast between the haplotype carrying the selected allele, which extends over a long chromosomal segment with high homozygosity, and the ancestral haplotypes, which show normal levels of variation. This asymmetry in haplotype structure is the basis of extended haplotype homozygosity (EHH) tests, which detect selection by comparing the length of homozygous haplotypes around a candidate allele to the genome-wide expectation.4, 5
Detection methods
Tajima's D, introduced in 1989, compares two estimators of the population mutation rate: one based on the number of segregating sites and one based on the average number of pairwise differences. Under neutrality and demographic equilibrium, these estimators are expected to be equal, yielding a Tajima's D of approximately zero. A selective sweep reduces pairwise diversity more than the number of segregating sites, producing a negative Tajima's D in the swept region. However, population expansion also produces negative Tajima's D values, making this statistic alone insufficient to distinguish between demographic and selective explanations.3
Fay and Wu's H statistic, developed in 2000, was specifically designed to detect the excess of high-frequency derived alleles that hitchhiking produces. When a beneficial allele sweeps to fixation, it carries derived variants at linked sites to high frequency, creating a distinctive skew in the derived allele frequency spectrum that is not produced by population expansion or other demographic processes. Fay and Wu demonstrated that the H statistic has substantially greater power to detect completed sweeps than Tajima's D, particularly when an outgroup sequence is available to polarise alleles as ancestral or derived.2
The extended haplotype homozygosity (EHH) approach, introduced by Sabeti and colleagues in 2002, exploits the fact that a selected allele that has risen rapidly in frequency will be embedded in an unusually long haplotype because recombination has not had time to break down the linkage with flanking markers. The integrated haplotype score (iHS), a standardised version of the EHH test developed by Voight and colleagues in 2006, compares the haplotype lengths around the ancestral and derived alleles at each SNP across the genome and identifies loci where one allele sits on an anomalously extended haplotype, indicative of recent positive selection.4, 5
The McDonald-Kreitman test takes a different approach by comparing the ratio of nonsynonymous to synonymous variation within species (polymorphism) to the ratio between species (divergence). Under neutrality, these ratios should be equal. An excess of nonsynonymous divergence relative to polymorphism indicates that positive selection has driven amino acid substitutions to fixation faster than expected under drift alone. This test is less sensitive to the confounding effects of demography than frequency-spectrum-based methods and has been widely applied to identify genes that have experienced recurrent adaptive evolution.17, 10
Classic examples
The lactase persistence allele provides one of the most thoroughly characterised examples of genetic hitchhiking in the human genome. In most mammals, the enzyme lactase, which digests the milk sugar lactose, is downregulated after weaning. In populations with a long history of dairying, however, mutations that maintain lactase expression into adulthood have risen to high frequency under strong positive selection. Enattah and colleagues identified the C/T-13910 variant upstream of the LCT gene as the primary allele conferring lactase persistence in Europeans.8 The selective sweep around this variant has produced one of the longest haplotypes in the human genome: Voight and colleagues reported that the lactase persistence allele shows an extremely high integrated haplotype score, indicating that a haplotype extending over several hundred kilobases has been swept to high frequency in Europeans within the last 5,000 to 10,000 years.5 Tishkoff and colleagues demonstrated that independent lactase persistence mutations arose in East African pastoralist populations, representing convergent adaptation with distinct hitchhiking footprints from the European variant.9
The SLC24A5 gene, which encodes a cation exchanger involved in melanin synthesis, provides another striking example. Lamason and colleagues identified a threonine-to-alanine substitution (A111T) in SLC24A5 that accounts for a substantial fraction of the difference in skin pigmentation between European and African populations. This allele is nearly fixed in Europeans but rare in African and East Asian populations. Genomic analyses reveal a classic hard-sweep signature around SLC24A5: dramatically reduced heterozygosity, extended haplotype homozygosity, and a strongly negative Tajima's D, consistent with recent and powerful positive selection.7, 18
In Drosophila melanogaster, which served as the original model system for hitchhiking research, genome-wide surveys have identified numerous regions of reduced diversity flanking positively selected loci. Bustamante and colleagues analysed patterns of polymorphism and divergence across thousands of genes and found evidence that a substantial fraction of amino acid substitutions in the Drosophila lineage have been driven by positive selection, each leaving hitchhiking footprints that collectively depress genome-wide diversity below the neutral expectation.10
Background selection as the inverse
Background selection, formalised by Charlesworth, Morgan, and Charlesworth in 1993, is the conceptual mirror image of genetic hitchhiking. Whereas hitchhiking describes the increase in frequency of neutral alleles linked to positively selected variants, background selection describes the decrease in frequency of neutral alleles linked to negatively selected (deleterious) variants. When purifying selection continually removes deleterious mutations from a population, it also removes the neutral variants that happen to be linked to those mutations on the same chromosomes, reducing the effective population size and the level of neutral diversity at linked sites.11
The genomic signatures of background selection and hitchhiking are difficult to distinguish in practice. Both processes reduce diversity in regions of low recombination, and both predict a positive correlation between recombination rate and nucleotide diversity, a pattern that has been widely observed in organisms from Drosophila to humans. Charlesworth and colleagues argued that background selection may be at least as important as hitchhiking in explaining the observed correlation between recombination and diversity, because deleterious mutations arise far more frequently than strongly beneficial ones and therefore exert a more pervasive, if individually weaker, effect on linked neutral variation.11, 14
Relationship to linkage disequilibrium
Genetic hitchhiking is fundamentally a consequence of linkage disequilibrium (LD), the non-random association of alleles at different loci on the same chromosome. In a freely recombining genome, alleles at different loci are statistically independent of one another, and selection at one site has no effect on allele frequencies at other sites. In reality, recombination rates vary across the genome, and loci that are physically close together on a chromosome tend to be inherited together more often than expected by chance, creating linkage disequilibrium that decays as a function of recombinational distance.6, 14
The HapMap project documented the fine-scale structure of linkage disequilibrium across the human genome, revealing that chromosomes are organised into discrete blocks of high LD separated by recombination hotspots where LD breaks down rapidly. Within these blocks, a selective sweep at any one site will affect the entire block, because there is insufficient recombination to uncouple linked neutral variants from the selected allele.6 The block structure of LD also means that the footprint of a selective sweep is not smoothly distributed but shows abrupt boundaries at recombination hotspots, a pattern that has been confirmed in empirical studies of sweep regions around genes such as LCT and SLC24A5.5, 6
In populations with reduced recombination, whether due to the absence of sexual reproduction (as in asexual organisms or mitochondrial genomes), chromosomal inversions that suppress crossing over, or regions near centromeres and telomeres where recombination is naturally low, hitchhiking effects are amplified. Slatkin and Wiehe extended the hitchhiking model to subdivided populations and showed that the reduction in diversity caused by a selective sweep is attenuated by population structure, because different subpopulations may carry the beneficial allele on different haplotypic backgrounds, mimicking a soft sweep even when the initial mutation was unique.16
Significance
Genetic hitchhiking has transformed the study of molecular evolution by providing a mechanistic link between natural selection at specific loci and the genome-wide patterns of variation observed in population genomic data. The recognition that positive selection leaves characteristic footprints in the form of reduced diversity, extended haplotypes, and skewed frequency spectra has enabled researchers to scan entire genomes for evidence of recent adaptation without any prior knowledge of which genes or phenotypes are under selection. Genome-wide scans for hitchhiking signatures have identified hundreds of candidate loci for recent positive selection in humans and other species, providing insights into the genetic basis of adaptation to novel diets, pathogens, climates, and other environmental challenges.5, 18
At the same time, the hitchhiking framework has highlighted the difficulty of distinguishing the effects of selection from those of demography. Population bottlenecks, founder effects, and admixture can all produce patterns in the frequency spectrum and haplotype structure that resemble selective sweeps, and distinguishing true hitchhiking signatures from demographic artefacts remains one of the central challenges of population genetics. The ongoing development of statistical methods that jointly model demography and selection, combined with the increasing availability of whole-genome sequence data from diverse populations, continues to refine the ability to detect and characterise hitchhiking events and to quantify the role of positive selection in shaping the genetic architecture of organisms.2, 13, 18
References
Soft sweeps: molecular population genetics of adaptation from standing genetic variation
Soft sweeps and beyond: understanding the patterns and probabilities of selection footprints under rapid adaptation