Overview
- Population genetics is the mathematical study of allele frequency changes within populations, providing the quantitative framework that unified Mendelian inheritance with Darwinian natural selection in the modern evolutionary synthesis.
- The Hardy-Weinberg principle establishes that allele frequencies remain constant in an idealized population absent selection, drift, mutation, migration, and non-random mating, serving as the null model against which all evolutionary change is measured.
- Foundational work by Fisher, Wright, and Haldane in the 1920s and 1930s demonstrated that even slight selective advantages could drive allele fixation over generations, while genetic drift plays a disproportionate role in small populations, and modern coalescent theory now enables reconstruction of population histories from genomic data.
Population genetics is the branch of biology that studies the distribution and change of allele frequencies within populations, and the mathematical forces that shape genetic variation over time. By applying statistical and mathematical models to the inheritance patterns first described by Gregor Mendel, population genetics provides the quantitative framework through which natural selection, genetic drift, mutation, and gene flow are understood as mechanisms of evolutionary change.2, 5 The field emerged in the early twentieth century as a resolution to the apparent conflict between Mendelian inheritance and Darwinian gradualism, and its theoretical foundations became the backbone of the modern evolutionary synthesis.
The Hardy-Weinberg principle
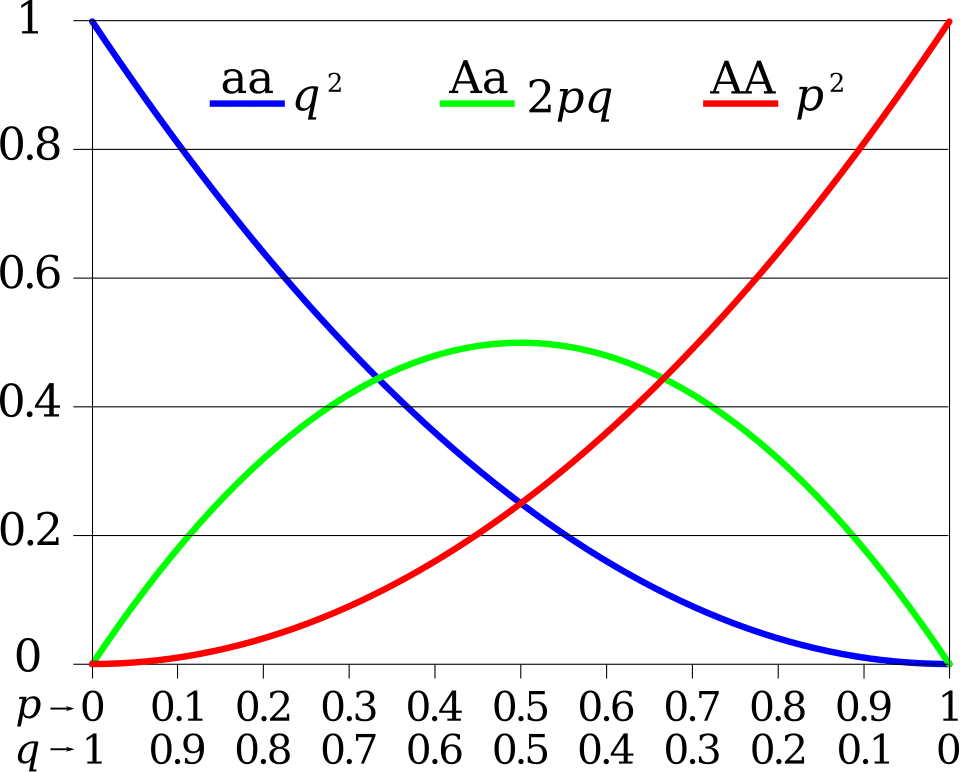
The foundational null model of population genetics was independently formulated in 1908 by the English mathematician G. H. Hardy and the German physician Wilhelm Weinberg.11, 12 The Hardy-Weinberg principle states that in an infinitely large, randomly mating population with no selection, no mutation, no migration, and no genetic drift, allele frequencies will remain constant from generation to generation, and genotype frequencies will settle into predictable proportions after a single generation of random mating.
{kind=link}
For a locus with two alleles at frequencies p and q (where p + q = 1), the expected genotype frequencies are p2, 2pq, and q2.10, 12
The principle's significance lies not in its assumptions, which are never perfectly met in nature, but in its function as a null hypothesis. Any deviation from Hardy-Weinberg equilibrium in a natural population indicates that one or more evolutionary forces are acting. Excess homozygosity, for example, may signal inbreeding or population subdivision, while a deficit of heterozygotes at a specific locus may indicate selection against heterozygous genotypes.10 The Hardy-Weinberg framework thus transformed evolutionary biology from a qualitative narrative into a quantitative science, providing a precise baseline against which the effects of selection, drift, mutation, and migration could be measured.
Fisher, Wright, and Haldane
The theoretical architecture of population genetics was constructed primarily by three mathematicians and biologists working in the 1920s and 1930s: Ronald A. Fisher, Sewall Wright, and J. B. S. Haldane. Their work, though sometimes conflicting in emphasis, collectively demonstrated that Mendelian genetics and Darwinian natural selection were not merely compatible but mutually reinforcing.2, 3, 4
Fisher's 1918 paper showed that continuous variation in quantitative traits, such as human height, could be explained by the combined effects of many Mendelian loci, each contributing small additive effects.1 This resolved the longstanding dispute between the biometricians, who studied continuous variation statistically, and the Mendelians, who emphasized discrete hereditary units. In his 1930 book The Genetical Theory of Natural Selection, Fisher formalized the mathematics of selection acting on allele frequencies in large populations and articulated the fundamental theorem of natural selection, which states that the rate of increase in mean fitness of a population equals its additive genetic variance in fitness.2, 17
Haldane, beginning with a landmark 1924 series of papers titled "A Mathematical Theory of Natural and Artificial Selection," provided the first rigorous calculations of how rapidly natural selection could change allele frequencies, demonstrating that even a selective advantage as small as one percent could drive an allele from rarity to near-fixation in several hundred generations.15 His calculations of the "cost of natural selection" — the total number of selective deaths required to substitute one allele for another — later became influential in debates over the relative importance of selection and drift at the molecular level.18
Wright developed a complementary framework emphasizing the role of population structure. His 1931 paper "Evolution in Mendelian Populations" introduced the concept of effective population size, the inbreeding coefficient, and the Wright-Fisher model of random genetic drift — a mathematical idealization in which a finite population of diploid individuals reproduces by random sampling of gametes each generation.3 Wright's shifting balance theory proposed that evolution proceeds most effectively when a species is subdivided into many small, partially isolated populations (demes), in which genetic drift can explore the adaptive landscape and discover novel fitness peaks that would be inaccessible to selection alone in a single large population.3, 13 Although the shifting balance theory remains debated, Wright's mathematical treatment of drift and population structure is universally accepted as foundational.
Population genetics and the modern synthesis
The theoretical work of Fisher, Wright, and Haldane provided the mathematical scaffolding upon which the modern evolutionary synthesis was built in the 1930s and 1940s. Theodosius Dobzhansky's 1937 book Genetics and the Origin of Species translated the abstract mathematical theory into the language of experimental genetics, using data from natural populations of Drosophila to demonstrate that the genetic variation predicted by population genetics theory actually existed in the wild and was maintained by balancing selection, drift, and migration.5

{kind=link}
The modern synthesis unified paleontology, systematics, and genetics under a common evolutionary framework in which population-genetic processes operating within species — microevolution — were understood to be sufficient, given time, to produce the large-scale patterns observed in the fossil record — macroevolution. Population genetics thus became the theoretical core of evolutionary biology, the discipline that connects heredity to evolutionary change at its most fundamental level.2, 5
The neutral theory and molecular evolution
In 1968, Motoo Kimura proposed the neutral theory of molecular evolution, which argued that the vast majority of evolutionary changes at the molecular level are caused not by natural selection but by the random fixation of selectively neutral or nearly neutral mutations through genetic drift.6 Kimura observed that the rate of amino acid substitution across proteins appeared remarkably constant over time — the molecular clock pattern — and argued that this constancy was difficult to reconcile with selection-driven substitution, which should vary with environmental conditions, but was a natural prediction of drift-driven fixation in populations of roughly constant effective size.6, 7
The neutral theory did not deny the importance of natural selection for adaptive evolution; rather, it proposed that most molecular variants are functionally equivalent and invisible to selection. In populations of effective size Ne, an allele with a selective coefficient s behaves effectively as neutral when the product Nes is much less than one — that is, when the force of drift overwhelms the force of selection.7, 18 The neutral theory stimulated decades of productive debate and provided the null model for modern molecular evolution, against which signatures of positive or purifying selection are now routinely tested using genomic data.14
Relative influence of evolutionary forces by effective population size7, 18
Coalescent theory
A major conceptual advance in population genetics came in the early 1980s with the development of coalescent theory by John Kingman.9
{kind=link}
Rather than tracing allele frequencies forward in time, as classical population genetics does, the coalescent traces the genealogy of a sample of alleles backward in time to their most recent common ancestor. In a Wright-Fisher population of effective size Ne, any two gene copies in the present generation share a common ancestor at an expected time of 2Ne generations in the past, and the genealogy of a larger sample forms a branching tree whose topology and branch lengths encode information about the population's demographic history.8, 9
Coalescent theory revolutionized the analysis of genetic data by providing efficient algorithms for simulating gene genealogies under complex demographic models, including population bottlenecks, expansions, subdivisions, and migration. It is now the standard framework for inferring population size changes, divergence times, and migration rates from DNA sequence data.8, 16 Methods such as the pairwise sequentially Markovian coalescent (PSMC) and its extensions can reconstruct the demographic history of a species from a single diploid genome, revealing population size fluctuations over hundreds of thousands of years. Application of these methods to human genomes has revealed multiple bottlenecks corresponding to glacial periods and the out-of-Africa migration.16
Population genetics in the genomic era
The availability of whole-genome sequence data from thousands of individuals across hundreds of species has transformed population genetics from a theory-heavy discipline into a data-rich science. Genome-wide scans for signatures of natural selection now routinely identify loci that have undergone recent adaptive evolution in human populations, including genes involved in lactase persistence, malaria resistance, skin pigmentation, and high-altitude adaptation.14 These studies rely on population-genetic predictions about the patterns that positive selection leaves in the genome: extended regions of reduced variation (selective sweeps), elevated linkage disequilibrium around the selected allele, and shifts in the allele frequency spectrum toward high-frequency derived variants.14
A powerful tool for distinguishing adaptive from neutral molecular evolution is the McDonald-Kreitman (MK) test, introduced by John McDonald and Martin Kreitman in 1991. The MK test compares the ratio of nonsynonymous (amino acid–altering) to synonymous (silent) substitutions fixed between species with the corresponding ratio of polymorphisms segregating within species. Under strict neutrality, these ratios should be equal; an excess of fixed nonsynonymous differences indicates positive selection, while an excess of nonsynonymous polymorphism indicates the segregation of mildly deleterious variants. Application of the MK test across the human genome has estimated that approximately 10 to 15 percent of amino acid substitutions in the human lineage since divergence from chimpanzees were driven by positive selection, with genes involved in transcription regulation, immunity, and reproduction showing the strongest signatures.20, 14
The most ambitious experimental test of population-genetic principles is Richard Lenski's Long-Term Evolution Experiment (LTEE), begun in 1988 with 12 initially identical populations of Escherichia coli. By 2024 the experiment had surpassed 80,000 bacterial generations, making it the longest-running evolution experiment in history. A landmark discovery came between generations 31,000 and 31,500 in one of the 12 populations, when cells evolved the ability to metabolize citrate under aerobic conditions—a trait absent in E. coli for its entire known evolutionary history. Blount, Borland, and Lenski showed that this innovation required the prior accumulation of specific "potentiating" mutations that occurred thousands of generations earlier, demonstrating the role of historical contingency in evolutionary innovation and illustrating how apparently "irreducibly complex" traits can arise through the stepwise accumulation of individually small genetic changes.19, 21
Population genomics has also deepened understanding of the interplay between selection and drift. In species with very large effective population sizes, such as bacteria and Drosophila, even weakly deleterious mutations are efficiently purged by purifying selection, and adaptive substitutions are relatively common. In species with small effective population sizes, such as humans and other large vertebrates, weakly deleterious mutations accumulate more readily because drift overpowers weak selection, a phenomenon that Kimura's theory predicted and that genomic data have now abundantly confirmed.7, 18 The ongoing synthesis of population-genetic theory with high-throughput genomic data continues to refine understanding of how evolution operates at the molecular level, connecting the abstract mathematics of allele frequency change to the concrete biological reality of adaptation, speciation, and the maintenance of genetic diversity.
References
Inferring human population size and separation history from multiple genome sequences
Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli