Overview
- Homo sapiens possesses remarkably low genetic diversity compared to other great apes, a consequence of a relatively recent origin in Africa followed by severe population bottlenecks during the Out-of-Africa expansion roughly 50,000 to 70,000 years ago.
- The vast majority of human genetic variation, approximately 85 to 95 percent, exists within populations rather than between them, and geographic distance from Africa is the single strongest predictor of a population's heterozygosity, consistent with a serial founder effect originating in sub-Saharan Africa.
- Although overall differentiation between continental groups is modest, natural selection has produced striking local adaptations such as lactase persistence, sickle cell resistance to malaria, and high-altitude tolerance, while archaic admixture from Neanderthals and Denisovans has contributed functionally important variation to non-African populations.
Human genetic diversity encompasses the full range of heritable variation present within and among populations of Homo sapiens. Despite inhabiting virtually every terrestrial biome on the planet, modern humans are a genetically homogeneous species: two randomly chosen individuals differ at only about 0.1 percent of their roughly 3.2 billion base pairs of nuclear DNA, and the overwhelming majority of that variation is shared across all populations rather than confined to particular geographic groups.2, 7 This low overall diversity, combined with a distinctive geographic pattern in which populations farther from Africa harbour progressively less variation, constitutes some of the strongest genetic evidence for a recent African origin of all modern humans. At the same time, the variation that does exist includes functionally significant adaptations to local environments — from disease resistance in the tropics to oxygen metabolism at high altitude — and carries the imprint of archaic admixture with Neanderthals and Denisovans. Understanding the patterns and origins of human genetic diversity is therefore fundamental to reconstructing our species' evolutionary history, to identifying the genetic basis of disease susceptibility, and to evaluating the biological validity of racial categories.3, 8
Low diversity among great apes
One of the most striking findings of comparative genomics is that Homo sapiens possesses remarkably low genetic diversity relative to its closest living relatives, the other great apes. A landmark 2013 study sequenced the genomes of 79 individuals representing all six great ape species — chimpanzees, bonobos, gorillas, and orangutans, in addition to humans — and found that human nucleotide diversity is among the lowest in the group.1 Central chimpanzees (Pan troglodytes troglodytes) and western lowland gorillas (Gorilla gorilla gorilla), both confined to relatively small ranges in equatorial Africa, harbour substantially more genetic variation than the entire global human population. Even individual chimpanzee subspecies, whose census populations number in the tens of thousands, can exceed the nucleotide diversity of a species that numbers eight billion.1
This deficit is not a consequence of the human genome being somehow intrinsically less variable; rather, it reflects the unusual demographic history of our species. Genomic and archaeological evidence converge on a picture in which anatomically modern humans evolved from a relatively small effective population in Africa, experienced one or more severe bottlenecks during the dispersal out of Africa beginning roughly 50,000 to 70,000 years ago, and only recently expanded to their current enormous census size.4, 6 Because genetic diversity accumulates slowly through mutation over many generations, the brief period since the global expansion has been insufficient to recover the variation lost during these bottlenecks. The contrast with chimpanzees is instructive: despite their far smaller census numbers, chimpanzee subspecies have been separated for hundreds of thousands of years, allowing each lineage to accumulate its own private variation independently.1
The Out-of-Africa bottleneck and serial founder effects
The geographic distribution of human genetic diversity preserves a remarkably clear signature of the species' migratory history. In 2005, Ramachandran and colleagues demonstrated that the heterozygosity of human populations — a standard measure of genetic diversity — declines in a nearly linear fashion as a function of geographic distance from East Africa, measured along plausible migration routes over land.4 Independently and simultaneously, Prugnolle, Manica, and Balloux reported the same pattern using a different set of microsatellite markers, confirming that geography is a better predictor of neutral genetic diversity than any cultural, linguistic, or ethnic classification.5
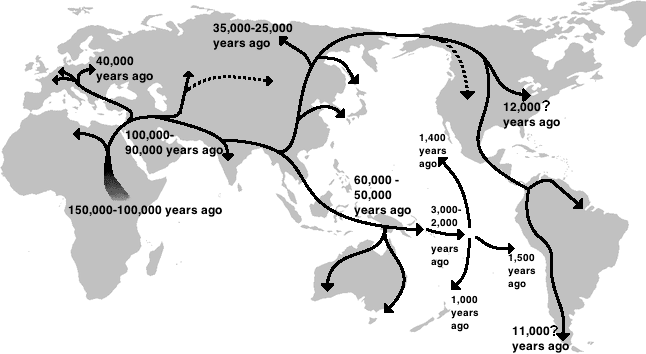
{kind=link}
This decline is best explained by a serial founder effect: as small groups at the frontier of the expanding human range splintered off and colonised new territory, each successive founding event sampled only a fraction of the genetic variation present in the parent population. Repeated over dozens of generations and thousands of kilometres, this process produced a smooth, cumulative erosion of diversity from Africa through the Middle East, Europe, and Asia, reaching its lowest point in the Americas and Oceania, the last major landmasses to be settled.4, 5 The 2008 analysis by Li and colleagues, which genotyped 650,000 single-nucleotide polymorphisms (SNPs) in 938 individuals from 51 populations of the Human Genome Diversity Panel, confirmed that haplotype heterozygosity declines smoothly with distance from Africa and that no geographic origin outside of Africa produces as good a fit to the observed data.6
Within Africa, by contrast, populations retain the deepest genetic lineages and the highest levels of diversity on Earth. The Bergström et al. 2020 analysis of 929 high-coverage genomes from the Human Genome Diversity Project (HGDP) identified an excess of previously undocumented common variants private to southern and central African populations, along with evidence for deep and gradual population separations within the continent extending back hundreds of thousands of years.3 These findings underscore that Africa is not merely the starting point of human migration but a reservoir of genetic complexity that remains incompletely characterised.
The 1000 Genomes and Human Genome Diversity Projects
Two large-scale sequencing efforts have provided the foundational catalogues of human genetic variation upon which most modern analyses depend. The 1000 Genomes Project, completed in 2015, reconstructed the genomes of 2,504 individuals from 26 populations across five continental regions using a combination of low-coverage whole-genome sequencing, deep exome sequencing, and dense microarray genotyping. The final data release characterised more than 88 million variants, including 84.7 million SNPs, 3.6 million short insertions and deletions (indels), and 60,000 structural variants, capturing over 99 percent of SNPs with a frequency above 1 percent in the sampled populations.2 A key finding was that while common variants are broadly shared across populations, rare variants — those with allele frequencies below 0.5 percent — tend to be geographically restricted, often found in only one or a few closely related populations.2
The Human Genome Diversity Project (HGDP), originally conceived by Luigi Luca Cavalli-Sforza in the 1990s to sample the full breadth of human population diversity, was transformed by modern sequencing technology. The 2020 analysis by Bergström and colleagues sequenced 929 individuals from 54 globally distributed populations to high coverage, identifying 67.3 million SNPs, 8.8 million indels, and 40,736 copy number variants.3 Critically, the HGDP includes populations that are underrepresented in the 1000 Genomes dataset, such as San and Mbuti hunter-gatherers from southern and central Africa, Aboriginal Australians, and several indigenous American groups, providing a more comprehensive picture of global variation. Together, these two resources have enabled researchers to construct detailed maps of human genetic diversity, identify signatures of natural selection, trace the history of admixture between archaic and modern human populations, and improve the resolution of genome-wide association studies for biomedical traits.2, 3
Population structure and FST
In 1972, the geneticist Richard Lewontin published a foundational analysis of human genetic variation using data from 17 polymorphic blood group and enzyme loci. He found that approximately 85 percent of total genetic variation existed among individuals within the same local population, roughly 8 percent differentiated populations within the same continental group, and only about 6 to 7 percent distinguished the major continental groups from one another.7 This result — that the great majority of human variation is shared, and that between-group differences account for a small fraction of the total — has been replicated with progressively larger and more precise datasets over the subsequent half century.20
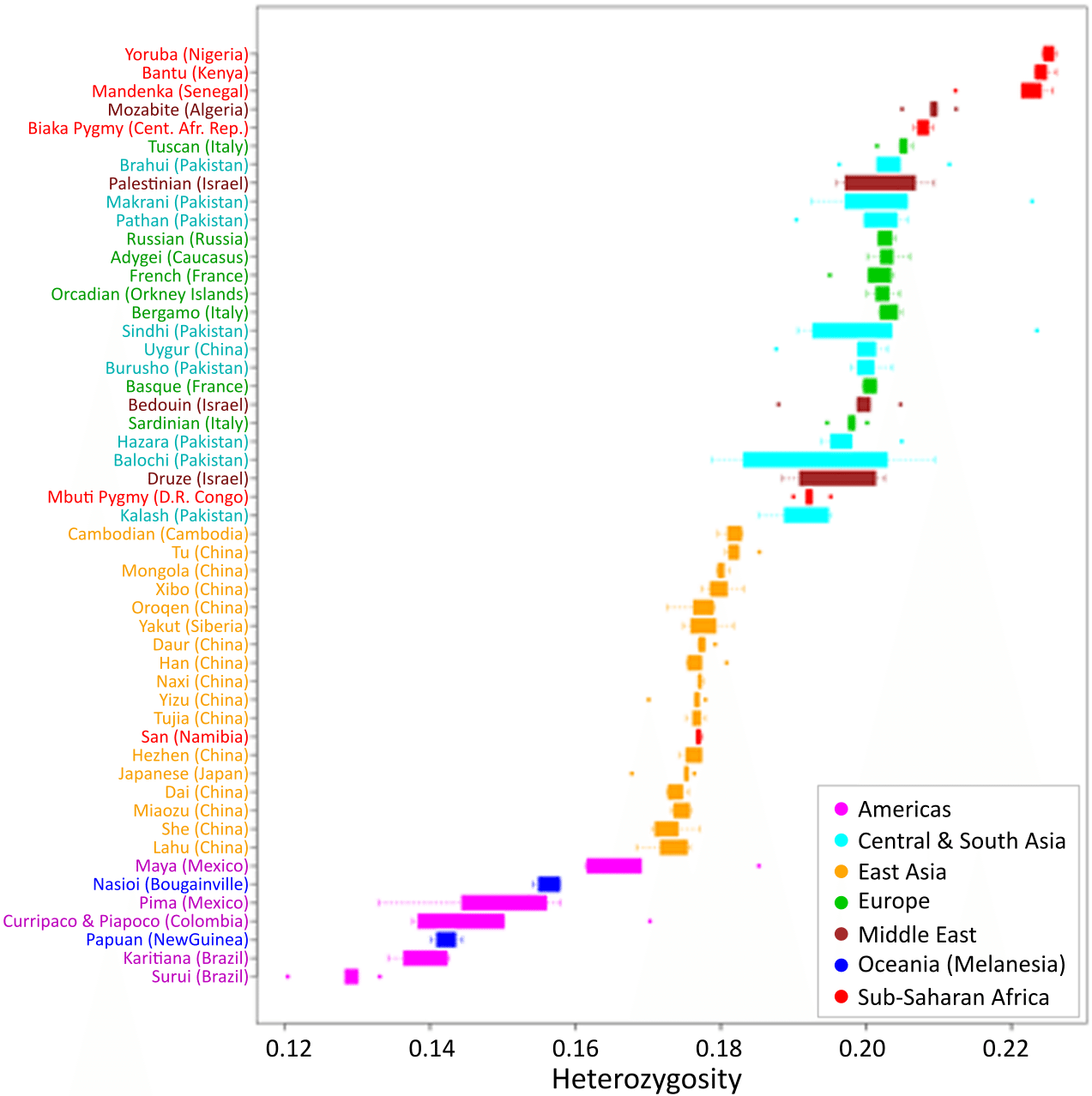
{kind=link}
The fixation index, FST, provides a formal measure of genetic differentiation between populations. FST values range from 0 (no differentiation; allele frequencies are identical) to 1 (complete differentiation; populations share no alleles). Global estimates of FST among human continental groups, computed from hundreds of thousands of genomic markers, consistently fall in the range of 0.05 to 0.15, depending on the markers and populations analysed.8, 21 By comparison, FST values between chimpanzee subspecies are substantially higher, often exceeding 0.30, reflecting their longer history of geographic separation and more restricted gene flow.1
In 2002, Rosenberg and colleagues used 377 microsatellite markers genotyped in 1,056 individuals from 52 populations to examine population structure using the model-based clustering algorithm STRUCTURE. Without any prior geographic information, the algorithm identified five major clusters that correspond broadly to Africa, Europe and the Middle East, East Asia, Oceania, and the Americas — but within-population variation accounted for 93 to 95 percent of the total, with between-group differences constituting only 3 to 5 percent.8 A follow-up study using 993 markers demonstrated that the apparent discreteness of these clusters is sensitive to study design: when populations are sampled evenly along geographic gradients rather than from widely separated regions, the pattern of variation appears as smooth clines rather than sharp boundaries, with the continental clusters emerging from the relatively thin strips of reduced habitation (oceans, deserts, mountain ranges) that separate major landmasses.9
Global FST values between continental population groups8, 21
The chart illustrates the modest magnitude of genetic differentiation between major human population groups. Even the largest pairwise FST values, between East Asian and sub-Saharan African populations, remain well below the threshold of 0.25 that population geneticists conventionally use to define "great" differentiation, and far below the values observed between subspecies of chimpanzees or gorillas.1, 8, 21
Functional variation and local adaptation
Although overall genetic differentiation between human populations is small, natural selection has produced a number of striking adaptations to local environmental conditions. These cases of functional variation demonstrate that even modest allele frequency differences can have significant phenotypic consequences when they occur in genes under strong selective pressure.
Lactase persistence is the ability to digest lactose, the primary sugar in milk, into adulthood. In most mammals, the enzyme lactase is downregulated after weaning, but in several human populations with a long history of pastoralism and dairy consumption, genetic variants near the lactase gene (LCT) maintain its expression throughout life. In European populations, a single SNP (C/T-13910) upstream of LCT is strongly associated with lactase persistence and shows evidence of a selective sweep within the past 5,000 to 10,000 years.10 In East African pastoralist populations, Tishkoff and colleagues identified three different SNPs (G/C-14010, T/G-13915, and C/G-13907) that independently produce the same phenotype, arising on distinct haplotype backgrounds from the European variant. This convergent evolution — different mutations in different populations producing the same adaptive trait — constitutes one of the most compelling examples of recent positive selection in the human genome.10
Resistance to malaria has exerted among the strongest selective pressures on the human genome in regions where Plasmodium falciparum is endemic. In 1954, Anthony Allison demonstrated that individuals heterozygous for the sickle cell allele (HbS) of the beta-globin gene experience substantially reduced mortality from severe malaria compared to individuals with two copies of the normal allele.11 The trade-off is that homozygotes for HbS develop sickle cell disease, a severe and often fatal condition. This balanced polymorphism maintains the HbS allele at frequencies of approximately 10 to 20 percent across much of tropical Africa, the Middle East, and parts of South Asia, with its geographic distribution closely mirroring the historical range of falciparum malaria.11, 12 The HbS variant is only one of several genetic defences against malaria; others include haemoglobin C, haemoglobin E, glucose-6-phosphate dehydrogenase (G6PD) deficiency, and the Duffy-negative blood group, each representing an independent evolutionary response to the same selective agent.19
High-altitude adaptation provides a third example. Populations living above 4,000 metres on the Tibetan Plateau, the Andean Altiplano, and the Ethiopian highlands have evolved distinct physiological strategies for coping with chronic hypoxia. In Tibetans, the strongest signal of natural selection in the entire genome occurs at EPAS1, a gene encoding a transcription factor in the hypoxia-inducible factor (HIF) pathway. A variant of EPAS1 found at approximately 78 percent frequency in Tibetans but only 9 percent in lowland Han Chinese is associated with lower haemoglobin concentrations at altitude, preventing the excessive polycythaemia (overproduction of red blood cells) that can cause complications such as chronic mountain sickness.13 Remarkably, the adaptive EPAS1 haplotype in Tibetans was acquired through introgression from Denisovans or a closely related archaic human population, representing a case in which archaic admixture provided the raw material for adaptation to a novel environment.14
Genetic variation and the concept of race
The distribution of human genetic variation has profound implications for evaluating the biological validity of racial categories. Traditional racial classifications, which sort humans into a small number of discrete groups on the basis of visible physical traits such as skin colour, hair form, and facial features, imply that the major divisions of humanity are sharply bounded, internally homogeneous, and genetically fundamental. The genomic evidence does not support this characterisation.7, 18
Lewontin's 1972 finding that 85 percent of human variation exists within rather than between conventionally defined racial groups has been confirmed by every subsequent genome-wide study.7, 20 Rosenberg and colleagues showed that the genetic clusters identifiable in global datasets correspond broadly to continental regions but are not sharp boundaries: they arise primarily from the geographic barriers (oceans, deserts) that have reduced gene flow between continents, and they dissolve into smooth clines when populations along geographic gradients are sampled more densely.8, 9 The HGDP analysis further demonstrated that common variants are shared across all major geographic regions and that no fixed genetic differences distinguish one continental population from another.3
In a formal population-genetic analysis, Templeton applied two standard biological definitions of "race" (or "subspecies") — one based on FST thresholds and one based on phylogenetic distinctiveness — to both human and chimpanzee genetic data. Chimpanzees satisfied the criteria for the existence of biological races under both definitions; humans did not. Human FST values are too low, and human population phylogenies are too reticulate (interconnected by gene flow) to meet the quantitative standards applied to other species.18 This does not mean that geographic ancestry is genetically undetectable — it clearly is, given sufficient markers — but it does mean that the pattern of human variation is one of gradients and overlapping distributions rather than discrete, non-overlapping types. Human genetic variation is real and geographically structured, but it does not conform to the typological categories implied by the concept of biological race.18, 21
Archaic admixture and its legacy
A significant component of genetic diversity in non-African populations derives not from new mutations or drift within Homo sapiens but from interbreeding with archaic human species encountered during the Out-of-Africa dispersal. The sequencing of the Neanderthal genome in 2010 by Green and colleagues revealed that all non-African human populations carry approximately 1 to 4 percent Neanderthal-derived DNA, consistent with admixture occurring in the Middle East or western Asia shortly after the initial exodus from Africa.15
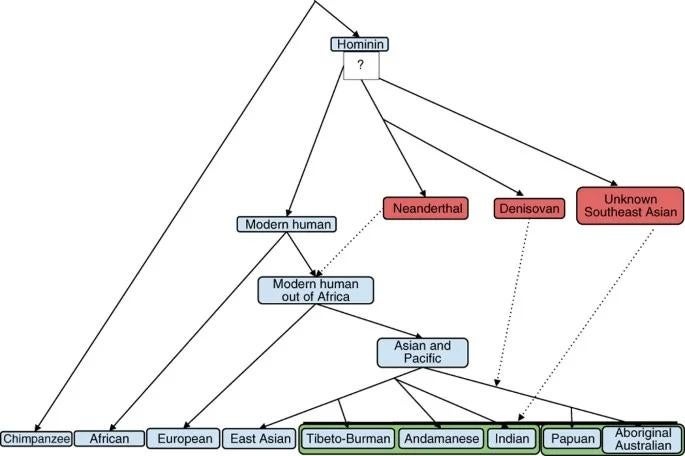
{kind=link}
Subsequent high-resolution analyses by Sankararaman et al. and Vernot and Akey mapped the distribution of Neanderthal ancestry across the modern human genome and found that it is not randomly distributed. Certain genomic regions are enriched in Neanderthal alleles, particularly those involved in keratin filament biology and skin pigmentation, suggesting that introgressed variants helped modern humans adapt to the colder, less sun-exposed environments of Eurasia.16, 17 The gene BNC2, associated with skin pigmentation variation in Europeans, carries an archaic haplotype present at approximately 70 percent frequency, representing one of the strongest candidates for adaptive introgression.17 Neanderthal-derived variants have also been linked to immune function, with alleles at several innate immunity loci (including Toll-like receptor genes) showing signatures of positive selection in Eurasian populations.22
Conversely, other regions of the genome show a pronounced depletion of Neanderthal ancestry, implying that natural selection has actively removed archaic alleles that were deleterious on a modern human genetic background. Genes expressed in the testes are especially depleted of Neanderthal variants, and the X chromosome carries approximately fivefold less Neanderthal ancestry than the autosomes, a pattern consistent with reduced male fertility in hybrids — a hallmark of incipient reproductive isolation between species.16 Neanderthal-derived variants have also been associated with increased risk for certain diseases, including type 2 diabetes, lupus, and Crohn's disease, through genome-wide association studies, although the effect sizes are typically small.23
Denisovan admixture adds a further layer of complexity. Present-day Melanesian populations carry approximately 3 to 6 percent Denisovan-derived DNA, and smaller but detectable Denisovan contributions have been identified in East Asian and South Asian populations.3, 15 The HGDP analysis provided evidence for multiple distinct Denisovan source populations contributing to present-day human genomes, in contrast to the apparently single Neanderthal source.3 The most celebrated example of adaptive Denisovan introgression is the EPAS1 high-altitude haplotype in Tibetans described above, which demonstrates that archaic admixture can provide adaptive variation that would have been unavailable from standing variation within the modern human gene pool.14
Biomedical and ethical implications
The patterns of human genetic diversity have direct consequences for biomedical research and clinical practice. Genome-wide association studies (GWAS), which identify genetic variants correlated with disease risk, have been conducted overwhelmingly in populations of European descent. As of the mid-2020s, individuals of European ancestry account for a disproportionate share of GWAS participants, despite representing a fraction of global genetic diversity.2, 3 Because allele frequencies, linkage disequilibrium patterns, and environmental exposures differ among populations, risk scores derived from European-ancestry GWAS may perform poorly when applied to individuals of African, Asian, or Indigenous American ancestry, potentially exacerbating rather than reducing health disparities.2
The catalogues generated by the 1000 Genomes Project and the HGDP have begun to address this imbalance by providing reference panels for imputation and analysis in diverse populations. The identification of previously undocumented common variants in underrepresented African and Oceanian populations underscores how much biomedically relevant variation remains to be characterised.3 Efforts to diversify the populations included in genomic research are not merely a matter of equity but a scientific necessity: the genetic architecture of complex diseases cannot be fully understood from the study of any single population, and some of the most medically important variants — such as the malaria-resistance alleles — are found predominantly or exclusively in populations that have been historically undersampled.12, 19
At the same time, the study of human genetic diversity raises ethical questions about privacy, group consent, and the potential for misuse of genetic information. The history of the original Human Genome Diversity Project, which faced criticism from indigenous communities concerned about the exploitation of their genetic material, illustrates the importance of conducting population-genetic research within frameworks of informed consent, community engagement, and equitable benefit-sharing.3 As sequencing costs continue to fall and genomic datasets grow, these ethical considerations will only become more pressing, requiring ongoing dialogue between researchers, policymakers, and the communities whose diversity is being studied.
References
Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa
Clines, clusters, and the effect of study design on the inference of human population structure
Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis
A population-genetic perspective on the similarities and differences among worldwide human populations