Overview
- The human genome is roughly 98% non-protein-coding sequence, a proportion explained by evolutionary processes including transposable element accumulation, pseudogenization, and neutral drift rather than by design.
- The ENCODE consortium's 2012 claim that 80% of the human genome is "functional" conflates biochemical activity with biological function; evolutionary geneticists showed that the vast majority of non-coding DNA evolves under no detectable purifying selection and can be lost without consequence.
- The C-value paradox — the observation that genome size varies enormously and unpredictably across species — is fatal to any argument that genome complexity reflects design; an onion has five times more DNA per cell than a human.
When the human genome was sequenced at the turn of the twenty-first century, one of its most striking revelations was how little of it encodes protein. Of the approximately three billion base pairs in the haploid human genome, fewer than two percent are grouped into the roughly 20,000 protein-coding genes that carry instructions for building the body’s machinery.6 The remaining 98% — encompassing introns, pseudogenes, transposable element remnants, satellite repeats, and vast stretches of sequence with no known role — has been called, since the early 1970s, junk DNA. That label has proven to be among the most contested in modern biology, generating a sustained scientific debate that acquired an unexpected ideological dimension when creationists and proponents of intelligent design seized upon a controversial 2012 study to argue that the genome contains no junk at all. Understanding what the evidence actually shows requires separating three distinct questions: what non-coding DNA is, whether it does anything, and whether whatever it does implies a designing intelligence.
{kind=link}
The origin of the junk DNA concept
The term junk DNA was coined by the geneticist Susumu Ohno in a 1972 paper that grappled with a puzzling discrepancy: if natural selection is efficient at eliminating harmful sequence and refining useful sequence, why do eukaryotic genomes contain so much more DNA than seems necessary for their protein-coding functions?1 Ohno proposed that the excess DNA was largely non-functional sequence accumulating by mutation and tolerated because it imposes little cost. He framed this as a consequence of evolution, not a flaw in it: the same relaxed selection that allowed pseudogenes — broken copies of formerly functional genes — to accumulate mutations also permitted non-coding sequence to drift without consequence.
In 1980, Leslie Orgel and Francis Crick extended this reasoning in a highly influential paper introducing the concept of selfish DNA.2 Orgel and Crick argued that many repeated elements in eukaryotic genomes are best understood not as functional components of the organism but as parasitic or quasi-parasitic sequences that persist because they are good at copying themselves. Natural selection operates on the organism as a whole, and if a sequence can replicate without imposing a sufficiently large fitness cost, it can spread through the population and persist indefinitely — regardless of whether it provides any benefit to its host. This framing, now supported by decades of molecular evidence, treats the genome not as a tightly optimized blueprint but as a palimpsest: a document written, overwritten, and cluttered with the evolutionary debris of hundreds of millions of years.
What non-coding DNA actually contains
The genome is not simply divided into genes and blank space. Non-coding sequence is itself heterogeneous, comprising several distinct classes of element with very different evolutionary histories and functional profiles.6, 8
Transposable elements are mobile genetic sequences that can copy themselves and insert elsewhere in the genome. They constitute at least 45% of the human genome by conservative estimates, and some analyses place the figure above two-thirds when divergent ancient copies are included.8 The major classes include SINEs (short interspersed nuclear elements, of which the Alu family is the most abundant human example), LINEs (long interspersed nuclear elements, particularly LINE-1), DNA transposons, and endogenous retroviruses — the fossilized remnants of retroviral infections that integrated into germline DNA and were inherited by all descendants. Most transposable element copies are ancient, heavily mutated, and incapable of further transposition; they are, in effect, molecular fossils. A small fraction remain active, and their occasional transposition generates mutations that can cause disease or, more rarely, produce novel regulatory sequences that are co-opted by evolution.11, 16
Pseudogenes are sequences that bear recognizable similarity to known protein-coding genes but have accumulated mutations — premature stop codons, frameshift insertions or deletions, disrupted splice sites — that prevent them from producing a functional protein.10 Processed pseudogenes arise when an mRNA is reverse-transcribed and reinserted into the genome, producing an intronless copy that typically lacks a functional promoter. Duplicated pseudogenes arise from gene duplication followed by degenerative mutation of one copy. The human genome contains thousands of pseudogenes, including a large repertoire of olfactory receptor pseudogenes, reflecting a dramatic reduction in functional olfactory receptor genes relative to other mammals. The pattern and distribution of shared pseudogenes across related species provides some of the strongest evidence for common descent, since the probability that two lineages would independently acquire identical disabling mutations at the same position approaches zero.
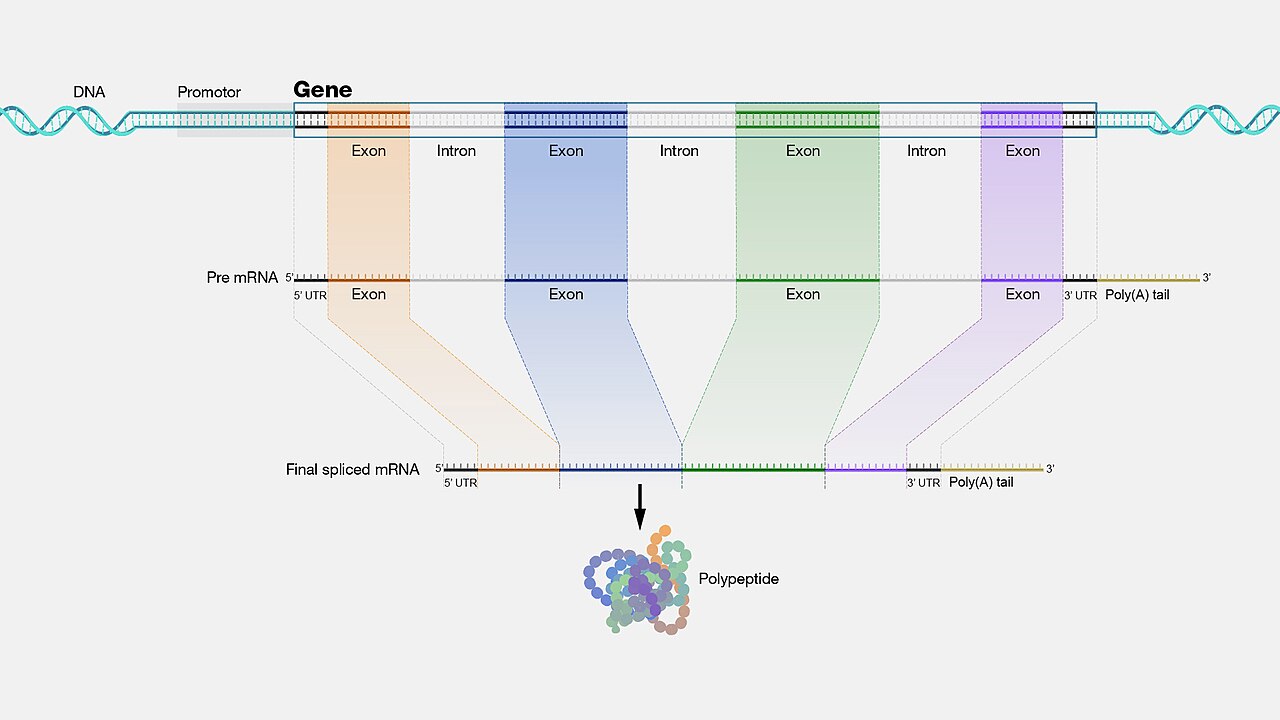
Introns are the non-coding intervening sequences that interrupt protein-coding genes and are excised from pre-mRNA before translation. Though introns are transcribed and therefore represent a substantial investment of cellular energy, the sequence of most intronic DNA evolves rapidly, suggesting that its specific content is not under strong selection. Splice sites and certain regulatory sequences within introns are conserved, but the bulk of intronic sequence appears to be largely free of functional constraint.6
Satellite DNA and tandem repeats consist of short sequence motifs repeated in tandem arrays at centromeres, telomeres, and other chromosomal regions. Centromeric and pericentromeric satellites are required for proper chromosome segregation during cell division, though the specific sequence of the repeating unit matters less than its presence; telomeric repeats protect chromosome ends from degradation and are essential for genome stability. Beyond these structural roles, most satellite DNA appears to be non-functional in any organismal sense.
Regulatory elements — enhancers, silencers, insulators, and promoter-distal control regions — are genuinely functional non-coding sequences that govern when, where, and at what level genes are expressed. They typically occupy only a small fraction of the genome, and while their discovery demonstrates that not all non-coding DNA is junk, their existence does not imply that the genome as a whole is functional. Identifying regulatory elements requires distinguishing them from the vast background of non-functional non-coding sequence, a distinction the ENCODE controversy repeatedly obscured.
The ENCODE controversy
In September 2012, the ENCODE (Encyclopedia of DNA Elements) Project Consortium published a landmark series of papers reporting the results of a large-scale survey of biochemical activity across the human genome.3 The project had assayed transcription factor binding, histone modification patterns, chromatin accessibility, and RNA transcription across dozens of cell types. Its headline finding — prominently announced in accompanying press releases and widely reported in major news outlets — was that 80% of the human genome is functional. The claim was repeated by scores of journalists and quickly entered popular discourse, where it was taken to mean that decades of evolutionary biology had been proven wrong and that the genome was far more purposefully organized than scientists had recognized.
The scientific response was swift and pointed. Dan Graur and colleagues published a detailed rebuttal in 2013 that identified what they described as a fundamental logical error at the heart of the ENCODE interpretation.4 The problem, they argued, was ENCODE’s conflation of two entirely different concepts: biochemical activity and biological function. A DNA sequence exhibits biochemical activity if it does something measurable in a biochemical assay — if a transcription factor binds to it, if it is transcribed into RNA, if it is marked by histone modifications associated with gene regulation. A sequence is biologically functional in the evolutionary sense if its loss or alteration would reduce organismal fitness — if natural selection is actively maintaining it against the erosive pressure of mutation. These two definitions are not equivalent.
Many sequences in the genome are biochemically active without being biologically functional. Transcription factors can bind to sequences adventitiously, simply because the sequences resemble their binding motifs by chance. RNA polymerase can produce low-level transcripts from genomic regions that lack functional significance — a phenomenon called transcriptional noise or pervasive transcription. Histone modifications can spread across large chromosomal domains without conferring specific regulatory function on every sequence within those domains. ENCODE detected all of these phenomena and counted them as evidence of function.4, 12
Evolutionary geneticist W. Ford Doolittle made the point with characteristic clarity: the ENCODE definition of function, if applied consistently, would also assign function to the transcription of sequences inserted by molecular biologists that are foreign to the human genome.12 A rigorously functional definition, he argued, must be grounded in the consequences of sequence variation for fitness. Graur and colleagues went further, noting that the ENCODE 80% figure is mathematically incompatible with what is known about human mutation rates and effective population sizes. For selection to maintain 80% of the genome against mutation, the number of deleterious mutations per generation would have to be so large that no lineage could sustain it — a constraint known as mutational load.4
Direct evolutionary analyses support this critique. Studies using comparative genomics — comparing aligned sequences across many mammalian species to identify regions evolving more slowly than the neutral rate, a hallmark of purifying selection — converge on estimates that between 5% and 15% of the human genome is under detectable constraint.5, 13 A 2014 analysis by Rands and colleagues, using constraint data from 35 mammalian genomes, estimated that approximately 8.2% of the human genome is subject to purifying selection; the remainder shows substitution rates indistinguishable from neutral sequence.5 Even this estimate likely includes some sequence conserved for structural rather than informational reasons, suggesting the true proportion of sequence with specific functional importance may be lower still.
The C-value paradox and the onion test
Long before the ENCODE controversy, biologists had identified a powerful argument against the idea that genome size tracks organismal complexity or design: the C-value paradox. The C-value is the total amount of DNA in a haploid genome, measured in picograms or base pairs. If genome size were a meaningful indicator of biological sophistication, one would expect complex organisms to have larger genomes than simpler ones. The data comprehensively contradict this expectation.9
Among flowering plants, genome size varies by a factor of more than 2,000, ranging from tiny genomes in plants like bladderwort (Utricularia gibba, which has pared its genome down to about 80 megabases) to enormous genomes in lilies and some ferns exceeding 100,000 megabases. Onions (Allium cepa) carry approximately 16,000 megabases per haploid cell — roughly five times the size of the human genome.9 Among salamanders, genome sizes vary by more than tenfold. Some lungfishes carry genomes more than forty times larger than the human genome. None of these differences track in any obvious way with the number of cell types, the complexity of body plans, or any other plausible measure of organismal sophistication.9, 14
Evolutionary biologist T. Ryan Gregory formalized this observation as the onion test: any claim that non-coding DNA is functional must explain why an onion needs five times more functional DNA per cell than a human being.14 The test is not a proof that junk DNA exists; it is a demand for a specific alternative explanation. Proponents of the view that all non-coding DNA is functional have not provided one. The most straightforward explanation for genome size variation across taxa is that most non-coding DNA is junk — specifically, that transposable element accumulation, which varies dramatically across lineages depending on effective population size and transposition rates, drives most of the variation in genome size.9 Smaller effective population sizes, as found in large-bodied organisms like mammals, reduce the efficiency of selection against slightly deleterious element insertions, allowing transposable elements to accumulate more readily — a prediction that matches observations across diverse taxa.
Evidence that most non-coding DNA is not functional
Beyond the C-value paradox, several independent lines of evidence converge on the conclusion that the majority of non-coding DNA is not under selection and is not biologically functional in the evolutionary sense.
Comparative conservation. If a sequence is functional, mutations that alter it will, on average, reduce fitness. Natural selection will therefore act against such mutations, slowing the rate of sequence evolution relative to neutral expectations. Conversely, sequence that evolves at the neutral rate is not under selection and is therefore not functional. Genome-wide surveys of constraint using multi-species alignments consistently show that the great majority of non-coding sequence in the human genome evolves at or near the neutral rate.5, 13 Regions of conserved non-coding sequence exist and are enriched for regulatory elements, but they account for a modest fraction of the total non-coding complement.
Deletion studies. In experimental organisms, large-scale deletions of non-coding sequence have been used to test directly whether that sequence is required for normal development and reproduction. Studies in mice have deleted hundreds of kilobases of apparently non-conserved non-coding sequence without producing detectable phenotypes, consistent with the interpretation that such sequence is dispensable.15 While the absence of an observed phenotype under laboratory conditions does not conclusively prove lack of function — subtle effects under natural conditions could be missed — the absence of phenotypic consequences in carefully designed deletion experiments is precisely the expected result if the deleted sequence is non-functional.
Mutation rate arguments. As Graur and colleagues detailed in their ENCODE rebuttal, the proportion of the genome maintained by selection can be estimated from first principles using knowledge of the per-generation mutation rate, effective population size, and the fraction of mutations that are deleterious.4 If 80% of the genome were functional, the number of new deleterious mutations entering the human population each generation would be orders of magnitude higher than what current estimates of mutation load can support. The argument implies an upper bound on the fraction of functional sequence that is consistent with the observed viability and fertility of human populations; that upper bound is far below 80%.
Genome streamlining. Some organisms face strong selection to minimize genome size, and in these lineages non-coding DNA is dramatically reduced. Parasitic plants and endosymbiotic bacteria with small effective population sizes and streamlined metabolisms have shed enormous amounts of non-coding sequence over evolutionary time, retaining only what is necessary for basic cellular function. The evolutionary lability of genome size across lineages — expanding readily when selection is relaxed and contracting when selection for compactness is intense — is exactly what is expected if non-coding DNA is largely neutral and can be gained or lost without major fitness consequences.9
Why the “no junk DNA” argument fails as evidence for design
Proponents of intelligent design have repeatedly invoked the ENCODE results, and more broadly the claim that junk DNA is a myth, as evidence that the genome was designed rather than evolved. The argument takes roughly the following form: evolutionary theory predicts junk DNA because evolution is a blind, undirected process that accumulates neutral or nearly neutral sequence; if the genome contains little or no junk, then evolution has been falsified and intelligent design is supported. This argument fails on multiple grounds.
First, as detailed above, the empirical claim that the genome contains little or no junk is not supported by the evidence. The ENCODE 80% figure reflects a specific definition of function that does not correspond to biological function in any evolutionary sense. The conclusion that most non-coding DNA is non-functional is supported by conservation analysis, deletion studies, mutational load calculations, and the C-value paradox. The intelligent design argument rests on a scientific claim that has been examined and found wanting by the relevant scientific community.4, 5, 12
Second, the argument misrepresents what evolutionary theory actually predicts. Evolutionary biology has never predicted that genomes should contain no functional non-coding sequence. Regulatory elements, non-coding RNAs with structural and catalytic roles, splice sites, replication origins, centromeric repeats, and many other categories of non-coding sequence have been recognized as functional for decades. The discovery that some fraction of non-coding DNA serves regulatory or structural roles is entirely consistent with evolutionary theory and was, in fact, predicted by it. What evolutionary theory does predict is that genomes will contain substantial amounts of sequence accumulated by neutral processes that is not maintained by selection — a prediction borne out by comparative genomics.
Third, and most fundamentally, the logic of the argument from design is structurally unsound. Even granting the premise that most non-coding DNA were functional, this would not constitute evidence for intelligent design over evolution. Natural selection acting over hundreds of millions of years is fully capable of co-opting, refining, and integrating non-coding sequence into regulatory networks. The gradual recruitment of transposable element sequences as cis-regulatory elements — documented in numerous lineages and providing concrete examples of evolutionary innovation arising from apparent junk — is a mechanism by which initially non-functional sequence can acquire function over time without invoking any directing intelligence.11, 16 The evolutionary track record of endogenous retroviruses being co-opted as regulatory elements controlling gene expression networks illustrates precisely this kind of repurposing.
Fourth, the argument from junk DNA, as presented in intelligent design literature, is formally an argument from ignorance: because we do not yet understand the function of a sequence, we should assume it has one, and that this assumed function implies design. This reasoning is problematic both scientifically and logically. The existence of sequence whose function is not yet characterized does not imply that such function exists; it implies only that more research is needed. As functional genomics has matured, the proportion of non-coding DNA with understood roles has grown, but the growth has not approached anything like the 80% claimed by ENCODE advocates, and the mechanisms by which function arises — gradual co-option of neutral sequence by selection — are entirely consistent with undirected evolutionary processes.
What is genuinely functional: a calibrated view
A scientifically calibrated account of genome function acknowledges both that a meaningful fraction of non-coding DNA is functional and that the majority is not. Regulatory elements — transcriptional enhancers, silencers, insulators, and the promoter-proximal sequences that govern gene expression — account for a significant portion of the conserved non-coding fraction and represent some of the most important sequence in the genome from a developmental and evolutionary standpoint.13 Comparative genomics across vertebrates has revealed hundreds of thousands of conserved non-coding elements, many of which have been experimentally validated as tissue-specific enhancers controlling the expression of developmental genes.
Non-coding RNA genes — producing ribosomal RNAs, transfer RNAs, small nuclear RNAs, microRNAs, long non-coding RNAs, and other species — represent another class of genuinely functional non-coding sequence. The roster of functionally characterized non-coding RNAs has expanded substantially since the mid-2000s, and some long non-coding RNAs have been implicated in processes including chromatin remodeling, dosage compensation, and genomic imprinting.6 However, functional annotation of a non-coding RNA requires the same rigor as for any other proposed functional element: evidence that the specific sequence is conserved and that its disruption produces fitness consequences, not merely that it is transcribed.
Even some transposable element sequences have been exapted — co-opted by natural selection for host functions distinct from the transposable element’s original replication strategy. Alu elements have contributed to gene regulation, and endogenous retroviral sequences have been recruited to control gene expression networks, including elements of the innate immune response.16 These examples of exaptation are genuine and scientifically significant; they demonstrate the creative potential of natural selection to repurpose existing sequence for new functions. They do not, however, imply that the average transposable element copy in the human genome is functional. The proportion of transposable element sequences that have been demonstrably exapted remains small relative to the total transposable element complement.
The emerging consensus — that roughly 5% to 15% of the human genome is under detectable purifying selection, with the remainder largely neutral — represents a mature synthesis of evolutionary theory and empirical genomics.5, 13 It preserves the core insight of Ohno’s original junk DNA hypothesis while refining the picture considerably: the genome is neither a perfectly optimized blueprint nor an undifferentiated sea of meaningless sequence, but a historically accumulated document in which genuine functional elements are embedded within a much larger background of evolutionary detritus. That picture is exactly what evolutionary theory predicts, and exactly what the evidence reveals.
References
On the immortality of television sets: 'function' in the human genome according to the evolution-free gospel of ENCODE
The C-value enigma in plants and animals: a review of parallels and an appeal for partnership
Identifying a high fraction of the human genome to be under selective constraint using GERP++