Overview
- Multiple independent lines of molecular evidence—DNA sequence similarity, shared pseudogenes, shared endogenous retroviruses, shared transposable element insertions, the universal genetic code, conserved synteny, and chromosome fusion—all converge on the same conclusion of universal common descent.
- Molecular phylogenies constructed from DNA and protein sequences independently recover the same branching tree as morphological and fossil evidence, and the degree of sequence divergence between species correlates with their estimated time of separation.
- This article serves as a hub connecting the detailed treatments of each molecular line of evidence, which together constitute arguably the most powerful category of evidence for evolution.
The molecular revolution in biology, beginning with the elucidation of DNA structure in 1953 and accelerating through the genomic era, has provided an entirely new category of evidence for common descent—one that is independent of, and concordant with, the anatomical, fossil, and biogeographic evidence that Darwin and his successors assembled.4 Every genome sequenced to date confirms the same fundamental finding: the degree of molecular similarity between organisms precisely tracks their evolutionary relatedness as inferred from other lines of evidence. DNA sequences, protein sequences, shared genomic accidents, chromosome architecture, and the genetic code itself all independently testify to the common ancestry of all life on Earth.3, 6
{kind=link}
This article provides an overview of the major molecular lines of evidence for common descent, each of which is treated in greater detail in dedicated articles linked throughout.
DNA and protein sequence similarity
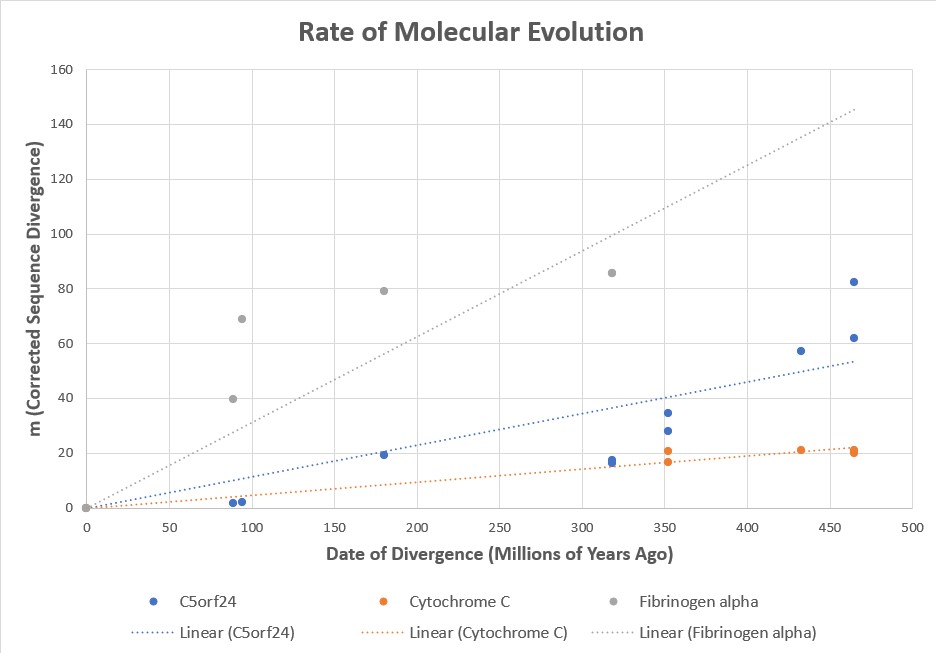
The most straightforward molecular evidence for common descent is the pattern of DNA and protein sequence similarity across species. When the genomes of humans and chimpanzees are compared, they share approximately 98.7% identity in aligned sequences, a figure that drops to roughly 96% when insertions and deletions are included.2 Human and gorilla genomes are slightly less similar; human and orangutan genomes less similar still; and the pattern continues outward through primates, other mammals, other vertebrates, and other animals, with decreasing similarity corresponding precisely to increasing evolutionary distance.
This pattern extends to individual proteins. Cytochrome c, an essential protein in the mitochondrial electron transport chain, has been sequenced in hundreds of species. Human cytochrome c is identical to chimpanzee cytochrome c at every amino acid position. It differs from the rhesus monkey version by one amino acid, from the dog version by eleven, from the tuna version by twenty-one, and from the yeast version by forty-four.5, 14 This nested pattern of similarity—in which more closely related organisms are more similar at the molecular level—is exactly what common descent predicts and has no coherent explanation under independent design.
Shared pseudogenes
Pseudogenes are broken, non-functional copies of genes that persist in the genome. When two species share the same pseudogene at the same chromosomal location, with the same inactivating mutations, the most parsimonious explanation is inheritance from a common ancestor in which the gene was broken. Humans and chimpanzees share thousands of pseudogenes at orthologous positions, each one carrying the same pattern of disabling mutations.8
The GULO pseudogene is a particularly well-studied example. Most mammals produce their own vitamin C via the enzyme L-gulonolactone oxidase, but humans carry a broken copy of the GULO gene with the same exon deletions and point mutations found in chimpanzees, gorillas, and other haplorrhine primates—indicating that the gene was broken once in a common ancestor approximately 40–65 million years ago. Other informative pseudogenes include the hundreds of shared olfactory receptor pseudogenes and the vitellogenin pseudogenes—remnants of egg yolk genes from egg-laying ancestors that persist as molecular fossils in the mammalian genome.8
Shared endogenous retroviruses
Endogenous retroviruses (ERVs) are the remnants of ancient retroviral infections that became permanently integrated into the germline DNA of ancestral organisms. Approximately 8% of the human genome consists of ERV sequences.1 Because retroviral integration into the host genome is essentially random with respect to position, the probability of independent insertions at the same chromosomal locus in two species is astronomically small. Humans and chimpanzees share thousands of ERV insertions at identical positions, each one a molecular fossil recording an infection that occurred in a shared common ancestor.7
The distribution of shared ERVs tracks primate phylogeny precisely. Insertions shared by humans and chimpanzees but absent from gorillas must have occurred after the gorilla lineage diverged; insertions shared by all great apes but absent from Old World monkeys must have occurred in the common ancestor of great apes. This phylogenetic concordance has been confirmed independently for multiple ERV families.7
Shared transposable element insertions
Transposable elements—including Alu elements, LINE-1 elements, and DNA transposons—constitute roughly 45% of the human genome.1 Like ERVs, these mobile DNA elements create unique molecular markers when they insert at specific chromosomal positions. Shared TE insertions at orthologous loci across species provide an independent line of evidence for common ancestry, based on the same statistical logic as shared ERVs but involving different classes of mobile DNA with different replication mechanisms.13
TE insertions are particularly valued as phylogenetic markers because they are essentially irreversible (precise excision is extremely rare) and virtually free from homoplasy (convergent independent insertion at the same site is negligibly probable). Shared SINE insertions have been used to resolve contested phylogenetic questions, including the placement of whales within artiodactyls and the branching order among placental mammal orders.13
Human chromosome 2 fusion
All great apes (chimpanzees, gorillas, orangutans) have 48 chromosomes (24 pairs), while humans have 46 (23 pairs). If humans share a common ancestor with the other great apes, a chromosome fusion event must have occurred in the human lineage, reducing the count by two. This prediction was confirmed when IJdo et al. discovered that human chromosome 2 contains the molecular signature of an ancient telomere-to-telomere fusion: a site in the middle of the chromosome (at band 2q13) where two blocks of telomeric repeat sequences (TTAGGG) are arranged head-to-head, exactly as would be expected if two ancestral chromosomes fused end-to-end.9
Further analysis revealed a vestigial centromere in the region corresponding to the centromere of one of the two ancestral chromosomes, now degenerated and non-functional.10 The genes flanking the fusion site on human chromosome 2 are found on two separate chromosomes in chimpanzees (chromosomes 2A and 2B), gorillas, and orangutans, confirming that the fusion occurred specifically in the human lineage after divergence from our last common ancestor with chimpanzees.9, 10
The universal genetic code
Virtually all life on Earth uses the same genetic code—the same mapping of 64 nucleotide triplets (codons) to 20 amino acids and stop signals. The probability that the same code would arise independently in multiple lineages by chance is vanishingly small; Freeland and Hurst calculated that the standard genetic code is more optimized for error minimization than all but approximately one in a million randomly generated alternative codes, yet there are over 1084 possible alternative codes.11 The universality of the code is most simply explained by inheritance from a single common ancestor in which the code was established.
Minor variations in the genetic code do exist—in mitochondria, certain ciliates, and a few other lineages—but these variations are small (typically involving the reassignment of one or two codons) and their phylogenetic distribution tracks evolutionary relatedness. Mitochondria in different lineages have independently acquired some of the same codon reassignments, consistent with the small number of viable modifications to a system constrained by its own complexity.11
Conserved synteny
Conserved synteny refers to the preservation of gene order along chromosomes across species. When the genomes of humans and mice are compared, large blocks of genes are found in the same order and orientation on corresponding chromosomes, despite approximately 90 million years of independent evolution since the last common ancestor of rodents and primates. Nadeau and Taylor first documented this pattern in 1984, showing that the mouse and human genomes could be decomposed into approximately 180 conserved syntenic segments that, when rearranged, would reconstruct something close to the ancestral mammalian genome.12
Conserved synteny is expected under common descent because chromosome rearrangements (inversions, translocations, fusions, fissions) accumulate gradually over evolutionary time. Closely related species share more syntenic blocks than distantly related ones, and the pattern of synteny breaks corresponds to the phylogenetic tree. Between humans and chimpanzees, synteny is nearly complete, with only a handful of chromosomal rearrangements (including the fusion of chromosome 2 discussed above) distinguishing the two genomes.2
Molecular phylogenetic concordance
Perhaps the most powerful aspect of the molecular evidence is that all of these independent data sources—DNA sequences, protein sequences, pseudogenes, ERVs, transposable elements, chromosome structure, and the genetic code—produce the same nested hierarchical pattern when used to reconstruct evolutionary relationships. Molecular phylogenies agree with morphological phylogenies, which agree with biogeographic distributions, which agree with the fossil record. Where disagreements exist, they are localized to specific nodes where rapid diversification makes resolution difficult, and they are resolvable with additional data.3, 6
Theobald formally tested the hypothesis of universal common ancestry against competing models of multiple independent origins and found that common ancestry was favored by a factor exceeding 102,860.3 This is not a marginal result. The molecular evidence for common descent is among the most thoroughly established conclusions in the history of science, supported by billions of data points from every genome ever sequenced, and challenged by no competing hypothesis that can account for the full pattern of molecular similarity, shared genomic accidents, and phylogenetic concordance observed across all of life.
References
Identification and analysis of unitary pseudogenes: historic and contemporary gene losses in humans and other primates
Genomic structure and evolution of the ancestral chromosome fusion site in 2q13–2q14.1 and paralogous regions on other human chromosomes