Overview
- Genes such as ASPM, Microcephalin, and FOXP2 show signatures of accelerated evolution along the human lineage, and mutations in these genes cause severe neurodevelopmental disorders, establishing direct links between specific loci and brain size, neural progenitor proliferation, and language-related cognition.
- Human accelerated regions (HARs) — genomic sequences that were deeply conserved across mammals but underwent rapid change specifically in the human lineage — and genome-wide studies of gene expression divergence between humans and chimpanzees indicate that regulatory evolution, not just protein-coding changes, has been a major driver of brain evolution.
- Human-specific gene duplications including SRGAP2C, ARHGAP11B, and NOTCH2NL have been shown to amplify neural progenitor populations and extend cortical neurogenesis, providing molecular mechanisms that help explain the threefold expansion of the human neocortex relative to the chimpanzee.
In 1975, Mary-Claire King and Allan Wilson demonstrated that human and chimpanzee proteins are more than 99% identical in amino acid sequence, despite the profound anatomical and cognitive differences between the two species.2 This finding posed a paradox that has shaped the genetics of brain evolution ever since: if the protein-coding genes of humans and chimpanzees are nearly indistinguishable, what genetic changes account for a brain that is three times larger, far more complexly folded, and capable of language, abstract reasoning, and cumulative culture? King and Wilson proposed that the answer lay not primarily in changes to the proteins themselves but in changes to the regulatory sequences that control when, where, and how much each gene is expressed during development.2 The fifty years of research since their landmark paper have confirmed the essential correctness of this insight while revealing a genetic landscape of human brain evolution that is richer and more varied than they could have anticipated — one that includes accelerated protein evolution at specific loci, non-coding regulatory changes, entirely new genes created by duplication, and sweeping alterations in the timing and magnitude of gene expression during cortical development.
The regulatory hypothesis
King and Wilson's 1975 paper in Science compared electrophoretic mobility, immunological cross-reactivity, and amino acid sequences of 44 proteins shared between humans and chimpanzees. They found an average sequence divergence of less than 1%, a degree of similarity exceeded by many pairs of sibling species within other mammalian genera.2 The contrast with the striking phenotypic differences between the two species was impossible to explain by protein evolution alone. King and Wilson therefore argued that the biological distance between humans and chimpanzees was primarily a consequence of mutations in regulatory elements — promoters, enhancers, and other non-coding sequences that govern gene expression — rather than mutations in the structural genes encoding proteins.2 This "regulatory hypothesis" predicted that the genetic basis of human-specific traits, including the enlarged brain, would be found predominantly in the non-coding genome. The completion of the chimpanzee genome sequence in 2005 confirmed the approximately 1.23% single-nucleotide divergence that King and Wilson had estimated, and revealed that indels (insertions and deletions) account for an additional 3% of sequence divergence, with many of the affected regions falling within or near regulatory elements rather than protein-coding exons.19
{kind=link}
ASPM and neural progenitor proliferation
Among the first genes to provide a direct link between a specific locus and human brain size was ASPM (Abnormal Spindle-like Microcephaly-associated). In 2002, Jacquelyn Bond and colleagues identified ASPM as the gene responsible for the most common form of autosomal recessive primary microcephaly (MCPH5), a condition in which the brain develops with a volume roughly one-third of normal despite otherwise largely normal architecture.1 The gene encodes a protein that localizes to the spindle poles of dividing neural progenitor cells and plays a critical role in symmetric cell division during cortical neurogenesis. When ASPM is mutated, neural progenitors undergo premature asymmetric division, depleting the progenitor pool and producing a dramatically smaller cortex.1
{kind=link}
The evolutionary significance of ASPM became apparent when Jianzhi Zhang demonstrated in 2003 that the gene had experienced a striking acceleration in its rate of amino acid substitution (elevated dN/dS ratio) along the primate lineage leading to humans.3 The signal of positive selection was concentrated in the IQ-repeat domains of the protein, which are thought to mediate interactions with calmodulin at the mitotic spindle. Across mammals, ASPM had evolved relatively slowly, but along the lineage from the last common ancestor of humans and chimpanzees to modern humans, the rate of non-synonymous substitution markedly exceeded the neutral expectation, indicating that natural selection had driven recurrent amino acid changes in the protein.3 This finding was among the earliest demonstrations that a gene with a known and direct role in determining brain size had been a target of positive selection specifically in the human lineage.
Microcephalin: selective sweeps and subsequent debate
A second primary microcephaly gene, Microcephalin (MCPH1), provided further evidence for the role of positive selection in human brain evolution. Andrew Jackson and colleagues cloned the gene in 2002, showing that loss-of-function mutations cause severe microcephaly with a brain volume reduced to approximately 400 cubic centimeters — comparable to that of an australopithecine.5 The encoded protein functions at the centrosome and is involved in DNA damage response and chromosome condensation during mitosis, processes essential for the orderly proliferation of neural progenitor cells.5
In 2005, Patrick Evans and colleagues reported that a specific allelic class of Microcephalin, designated haplogroup D, had undergone a strong selective sweep in human populations, rising to high frequency within the past approximately 37,000 years.4 The statistical signature of this sweep — extended linkage disequilibrium around the D allele, reduced nucleotide diversity on D-bearing chromosomes, and a frequency too high to be explained by neutral drift — led Evans and colleagues to argue that haplogroup D had been driven to prominence by positive natural selection, possibly related to brain size or brain function.4 A companion paper reported a similarly recent selective sweep at ASPM, with a positively selected allele estimated to have arisen approximately 5,800 years ago.4
These claims attracted immediate and vigorous scrutiny. Nicholas Timpson and colleagues published a direct test in 2007 using a large British cohort and found no association between the putatively selected alleles of Microcephalin or ASPM and any measure of brain size, head circumference, or general cognitive ability.6 The absence of a phenotypic effect in a well-powered sample cast doubt on the hypothesis that these recent selective sweeps were driven by effects on brain size, though it did not rule out selection on other phenotypes or selection on brain function in ways not captured by volume measurements. The episode illustrates a recurring theme in the genetics of brain evolution: identifying a gene under positive selection is far easier than identifying the specific phenotypic target of that selection.6
FOXP2 and the genetics of language
No gene has attracted more attention in the study of human cognitive evolution than FOXP2, the first gene to be directly linked to a specific aspect of human language. The gene came to attention through the KE family, a large multigenerational pedigree in which roughly half the members were affected by a severe speech and language disorder characterized by difficulty with the sequential coordination of mouth and face movements (orofacial dyspraxia) along with broader impairments in grammar, syntax, and linguistic comprehension.7 In 2001, Cecilia Lai and colleagues identified the genetic basis of the disorder as a point mutation in FOXP2, a gene encoding a transcription factor of the forkhead-box family. The mutation, an arginine-to-histidine substitution in the forkhead DNA-binding domain, abolished the protein's ability to regulate its downstream targets and segregated perfectly with the disorder in the KE family.7
The evolutionary analysis of FOXP2 followed swiftly. In 2002, Wolfgang Enard and colleagues sequenced the gene in humans, chimpanzees, gorillas, orangutans, rhesus macaques, and mice, finding that the protein is extraordinarily conserved across mammals: the mouse and human proteins differ at only three amino acid positions across 715 residues, and two of those three changes occurred on the human lineage after the divergence from chimpanzees.8 The two human-specific substitutions — a threonine-to-asparagine change at position 303 and an asparagine-to-serine change at position 325 — were accompanied by a pattern of flanking nucleotide variation consistent with a selective sweep that Enard and colleagues estimated had occurred within the last 200,000 years, roughly coinciding with the emergence of anatomically modern humans.8
Subsequent work revealed that FOXP2 functions as a transcription factor that regulates hundreds of downstream genes during brain development, many of them involved in neurite outgrowth, synaptogenesis, and the development of the cortico-basal ganglia circuits that control motor sequencing.22 Genevieve Konopka and colleagues demonstrated in 2009 that the human version of FOXP2 and the chimpanzee version regulate partly different sets of target genes in human neuronal cell lines, suggesting that the two human-specific amino acid changes altered the transcriptional program of the protein in ways relevant to neural circuit development.22 Enard and colleagues further showed that mice engineered to carry the humanized version of Foxp2 exhibited changes in dopamine levels and synaptic plasticity in the basal ganglia, along with altered ultrasonic vocalizations, demonstrating that the human-specific substitutions had detectable effects on neural circuitry in a model organism.9
The discovery that Neanderthal DNA preserves the same two derived amino acid substitutions found in modern human FOXP2 raised the question of whether the shared changes predate the split between the two lineages approximately 500,000–700,000 years ago rather than arising within the modern human lineage as originally proposed.20 The presence of the human-type FOXP2 in Neanderthals is consistent with, though does not prove, some degree of spoken language capacity in that species, and suggests that whatever selective advantage the human-specific substitutions conferred was established before the divergence of the Neanderthal and modern human lineages.
Human accelerated regions
While the studies of ASPM, Microcephalin, and FOXP2 identified individual protein-coding genes under positive selection, a complementary approach sought to identify rapidly evolving non-coding sequences across the entire genome — precisely the regulatory changes that King and Wilson had predicted. In 2006, Katherine Pollard and colleagues published two landmark papers that defined the concept of human accelerated regions (HARs): short genomic sequences that were deeply conserved across vertebrate or mammalian evolution but underwent a burst of nucleotide substitution specifically on the human lineage after divergence from the last common ancestor with chimpanzees.10, 11
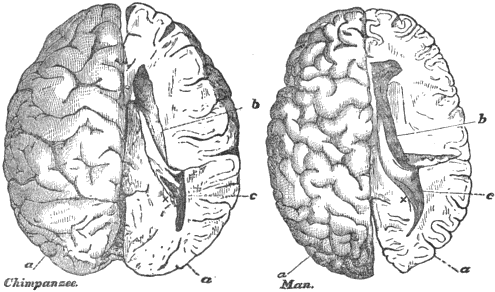
Pollard and colleagues identified 49 HARs by comparing the human genome to orthologous sequences in chimpanzees, mice, rats, and chickens, searching for regions that showed strong conservation across non-human vertebrates (indicating functional constraint) but an excess of human-specific substitutions relative to the neutral expectation.11 The most dramatically accelerated of these, designated HAR1, had accumulated 18 human-specific substitutions in a 118-base-pair segment that was otherwise nearly identical between chimpanzees and chickens — two species separated by approximately 310 million years of evolution.10 Functional characterization revealed that HAR1 is part of a novel RNA gene, HAR1F, that is expressed in Cajal-Retzius neurons of the developing human cortex between 7 and 19 gestational weeks, precisely the period during which these neurons guide the laminar organization of the cortical plate through the secretion of the signaling molecule reelin.10 The human and chimpanzee versions of the HAR1 RNA fold into different secondary structures, suggesting that the human-specific substitutions altered the function of this regulatory RNA in a way potentially relevant to cortical development.10
Independently, Shyam Prabhakar and colleagues identified a largely overlapping set of human-accelerated conserved non-coding sequences using a different computational method, confirming the robustness of the finding.21 Across the various studies, the majority of HARs fell in non-coding regions of the genome, consistent with King and Wilson's prediction that regulatory evolution would predominate. Many were located near genes involved in transcription regulation and neurodevelopment, and subsequent functional analyses have shown that a substantial fraction of HARs act as transcriptional enhancers active in the developing brain.10, 11, 21
Gene expression divergence
Beyond changes to individual genes or regulatory elements, genome-wide studies of gene expression have revealed that the human brain is distinguished from the chimpanzee brain by coordinated changes in the expression levels of hundreds of genes, with a pronounced bias toward upregulation in the human cortex. Philipp Khaitovich and colleagues compared gene expression profiles across multiple tissues in humans and chimpanzees using microarray technology, finding that the brain showed a higher degree of expression divergence than other tissues such as liver or heart, and that the direction of change was asymmetric: significantly more genes were upregulated in the human brain relative to the chimpanzee brain than were downregulated.13 The genes showing the strongest human-specific upregulation were enriched for functions related to synaptic transmission, energy metabolism, and signal transduction, suggesting that the human cortex has undergone a broad metabolic and synaptic intensification relative to the chimpanzee cortex.13
Mario Cáceres and colleagues extended these findings by focusing on the genes most dramatically overexpressed in the human cortex compared with the chimpanzee cortex. Among the most striking upregulations they identified were two members of the thrombospondin family, THBS2 and THBS4, which are secreted glycoproteins known to promote synaptogenesis — the formation of new synaptic connections between neurons.12 Both genes showed several-fold higher expression in the human cortex than in the chimpanzee cortex, while their expression in non-cortical tissues was similar between the two species. Cáceres and colleagues proposed that the cortex-specific upregulation of these synaptogenic factors may contribute to the higher density of synaptic connections in the human cortex, a feature that is likely to be at least as important for cognitive function as the overall increase in cortical volume.12
Human-specific gene duplications
Some of the most consequential genetic changes in human brain evolution involved not the modification of existing genes but the creation of entirely new ones through segmental duplication. In two companion papers published in Cell in 2012, Megan Dennis, Cécile Charrier, and colleagues described the evolutionary history and functional consequences of SRGAP2 duplication in the human lineage. The ancestral SRGAP2 gene encodes a signaling protein that promotes the maturation of dendritic spines — the small protrusions on neurons that receive synaptic inputs. Dennis and colleagues showed that the SRGAP2 locus was duplicated three times during hominin evolution: first approximately 3.4 million years ago (producing SRGAP2B), then approximately 2.4 million years ago (producing SRGAP2C), and finally approximately 1 million years ago (producing SRGAP2D).23 The timing of the second duplication, which produced the functionally active SRGAP2C paralog, coincides approximately with the transition from Australopithecus to early Homo and the onset of significant brain expansion in the hominin lineage.23
Charrier and colleagues demonstrated that SRGAP2C encodes a truncated protein that acts as a dominant negative inhibitor of the ancestral SRGAP2 protein. By blocking SRGAP2 function, SRGAP2C delays the maturation of dendritic spines, causing them to remain in a more juvenile, elongated state for an extended period during development — a form of neoteny at the cellular level.14 This delay in spine maturation increases the total density of synaptic spines on cortical neurons, potentially enhancing the capacity for synaptic connectivity. Mice engineered to express SRGAP2C displayed the same neotenous spine phenotype, with an increased density of immature spines resembling the pattern seen in human neurons.14
An even more dramatic example of duplication-driven brain evolution came from ARHGAP11B, a human-specific gene generated by a partial duplication of ARHGAP11A that occurred after the divergence of the human and chimpanzee lineages approximately 5 million years ago. In 2015, Marta Florio and colleagues showed that ARHGAP11B is expressed specifically in basal progenitor cells of the developing cortex — the transit-amplifying progenitors of the outer subventricular zone (OSVZ) that are massively expanded in the human brain compared with the brains of lissencephalic (smooth-brained) mammals.15 When ARHGAP11B was introduced into the developing mouse cortex, it caused a dramatic amplification of basal progenitors and, in some cases, the formation of cortical folds in the normally smooth mouse brain. The same effect was observed in the developing ferret cortex, where ARHGAP11B expression increased the number of basal radial glia and expanded the surface area of the cortex.15 These experiments provided some of the most compelling evidence that a single human-specific gene can produce tangible changes in brain size and cortical folding.
A third set of human-specific duplicated genes, the NOTCH2NL family, was identified in 2018 by Ian Fiddes and colleagues. These genes, located on chromosome 1 within a region associated with neurodevelopmental disorders when deleted or duplicated, arose through the partial duplication of NOTCH2 and are expressed in radial glia — the neural stem cells of the developing cortex.18 Fiddes and colleagues demonstrated that NOTCH2NL proteins enhance Notch signaling by acting as decoy ligands, delaying the differentiation of radial glia and thereby expanding the progenitor pool available for cortical neuron production. Deletion of the NOTCH2NL locus in human organoids led to smaller organoid size, while overexpression produced larger organoids with more cortical progenitors, establishing a direct link between these human-specific genes and cortical size regulation.18
Key genes implicated in human brain evolution1, 8, 15
| Gene | Function | Human-specific change | Key evidence |
|---|---|---|---|
| ASPM | Neural progenitor proliferation via spindle orientation | Accelerated amino acid evolution (elevated dN/dS) | Mutations cause primary microcephaly (MCPH5) |
| Microcephalin | DNA damage response, centrosomal function in progenitors | Selective sweep of haplogroup D | Loss-of-function causes microcephaly (MCPH1) |
| FOXP2 | Transcription factor regulating cortico-basal ganglia circuits | Two amino acid substitutions since chimpanzee divergence | Mutations cause severe speech and language disorder |
| HAR1F | Non-coding RNA in Cajal-Retzius neurons during cortical lamination | 18 substitutions in 118 bp (conserved across 310 Myr) | Expressed during critical cortical development window |
| SRGAP2C | Dominant-negative inhibitor of dendritic spine maturation | Gene duplication ~2.4 million years ago | Increases spine density, induces neoteny in mice |
| ARHGAP11B | Amplifies basal progenitors in the outer subventricular zone | Partial gene duplication unique to humans | Induces cortical folding in mouse and ferret |
| NOTCH2NL | Enhances Notch signaling to delay progenitor differentiation | Partial duplication of NOTCH2, human-specific | Deletion reduces organoid size; duplication enlarges it |
Outer radial glia and the expanded progenitor niche
The genes discussed above — ARHGAP11B, NOTCH2NL, and to some extent ASPM — converge on a common cellular mechanism: the amplification of neural progenitor populations in the developing cortex, particularly in the outer subventricular zone (OSVZ). The OSVZ is a germinal layer that is massively expanded in species with large, folded cortices, including humans and other primates, but is thin or absent in lissencephalic species such as mice.16 The dominant progenitor cell type of the OSVZ is the outer radial glial cell (oRG), also called a basal radial glial cell, which shares many molecular features with the ventricular radial glia of the ventricular zone but has detached from the ventricular surface and retains only a basal (pial-directed) process.16
{kind=link}
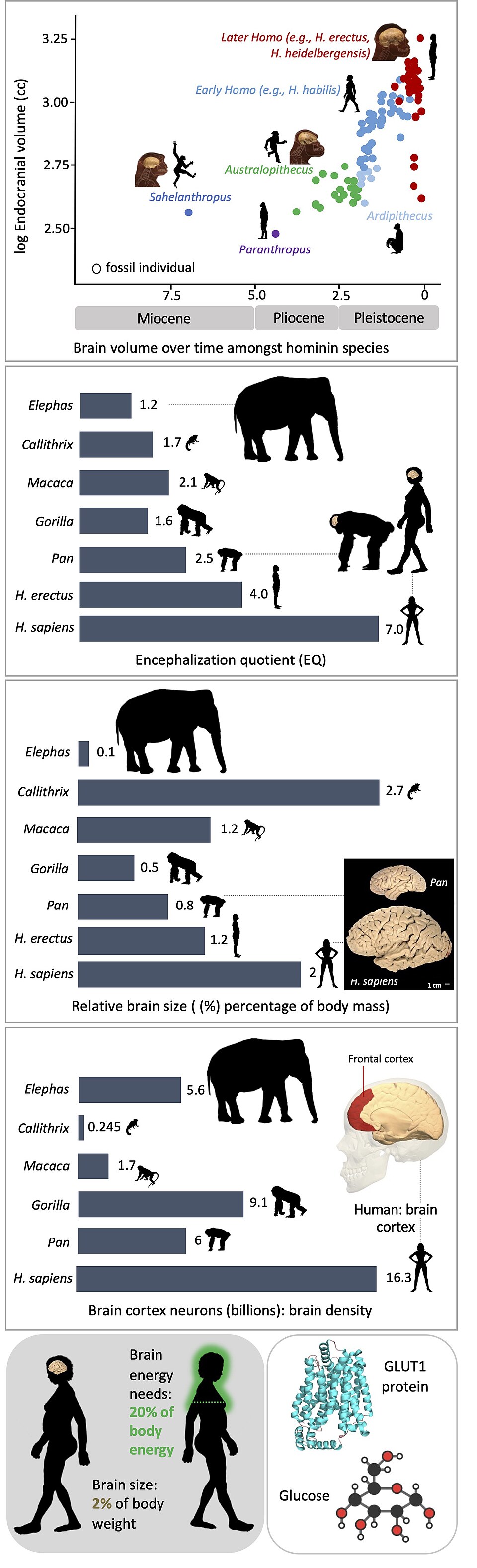
Jan Lui, David Hansen, and colleagues provided a comprehensive characterization of outer radial glia in 2011, demonstrating that these cells are self-renewing, highly proliferative neural stem cells that can generate large numbers of intermediate progenitor cells and, ultimately, cortical neurons.16 The abundance of outer radial glia in the human OSVZ, combined with their capacity for multiple rounds of symmetric self-renewing division before terminal differentiation, provides a cellular mechanism for the dramatic amplification of cortical neuron number that distinguishes the human brain from those of other mammals. In mice, the cortex is generated almost entirely from ventricular zone progenitors, and the total number of cortical neurons is roughly 14 million. In humans, the addition of the OSVZ progenitor pool expands this number to approximately 16 billion cortical neurons — a more than thousandfold increase that cannot be accounted for by the modest differences in ventricular zone progenitor number between the two species.16
The human-specific genes ARHGAP11B and NOTCH2NL both act within this progenitor niche. ARHGAP11B amplifies basal progenitors, increasing the pool of transit-amplifying cells that generate cortical neurons.15 NOTCH2NL delays the differentiation of radial glia, extending the period during which these stem cells can undergo self-renewing divisions and thereby increasing the total output of the progenitor compartment.18 Together, these and other human-specific genetic changes may have extended and amplified the neurogenic program of the developing cortex, producing the vast expansion of cortical surface area and neuron number that characterizes the human brain.
Enhancer evolution and the regulatory landscape
Returning to King and Wilson's original prediction, genome-wide studies of chromatin accessibility and enhancer activity have confirmed that regulatory evolution has been a pervasive force in shaping the developing human brain. Steven Reilly and colleagues published a landmark study in 2015 in which they used chromatin immunoprecipitation followed by sequencing (ChIP-seq) to map the activity of promoters and enhancers across the genome in developing human and rhesus macaque brains at matched developmental stages.17 They identified more than 2,800 promoters and enhancers that showed significantly different activity between the two species, with the majority of differences reflecting gains of enhancer activity on the human lineage rather than losses.17
These human-gained enhancers were disproportionately located near genes involved in cortical development, neural progenitor proliferation, and synaptic function, and many were active specifically during the fetal period of cortical neurogenesis.17 Reilly and colleagues further showed that human-gained enhancers were enriched in HARs and in regions of the genome that had experienced human-specific sequence changes, directly linking the accelerated sequence evolution detected by computational approaches to measurable changes in gene regulation during brain development.17 The study provided perhaps the most comprehensive empirical confirmation of King and Wilson's 1975 prediction: the genetic basis of human brain evolution is substantially regulatory, involving thousands of changes to the enhancers and promoters that sculpt the gene expression programs of the developing cortex.
The emerging picture is one in which human brain evolution was driven by a confluence of genetic mechanisms operating at multiple levels. A small number of protein-coding genes — ASPM, Microcephalin, FOXP2, and a handful of others — experienced accelerated amino acid evolution that altered their function in neural progenitor proliferation, cortical organization, or language-related neural circuits.1, 3, 8 Non-coding sequences that had been conserved for hundreds of millions of years underwent rapid change specifically on the human lineage, producing new regulatory RNAs and altered enhancer activities that rewired the gene expression programs of the developing brain.10, 17, 21 Entirely new genes arose through segmental duplication, providing novel molecular tools for amplifying neural progenitor populations and modulating synaptic connectivity.14, 15, 18 And pervasive changes in gene expression levels, affecting hundreds of genes simultaneously, recalibrated the metabolic, synaptic, and signaling properties of the human cortex relative to that of our closest relatives.12, 13 Together, these changes produced the organ that defines our species — not through a single genetic revolution but through the cumulative action of diverse molecular mechanisms, each contributing incrementally to the expansion, reorganization, and functional elaboration of the human brain.
References
Increased cortical expression of two synaptogenic thrombospondins in human brain evolution
Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation
Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion
Radial glia in the primate brain: neural stem cells, transitional forms, and species differences
Human-specific duplications of SRGAP2 occurred at key transition points in human evolution