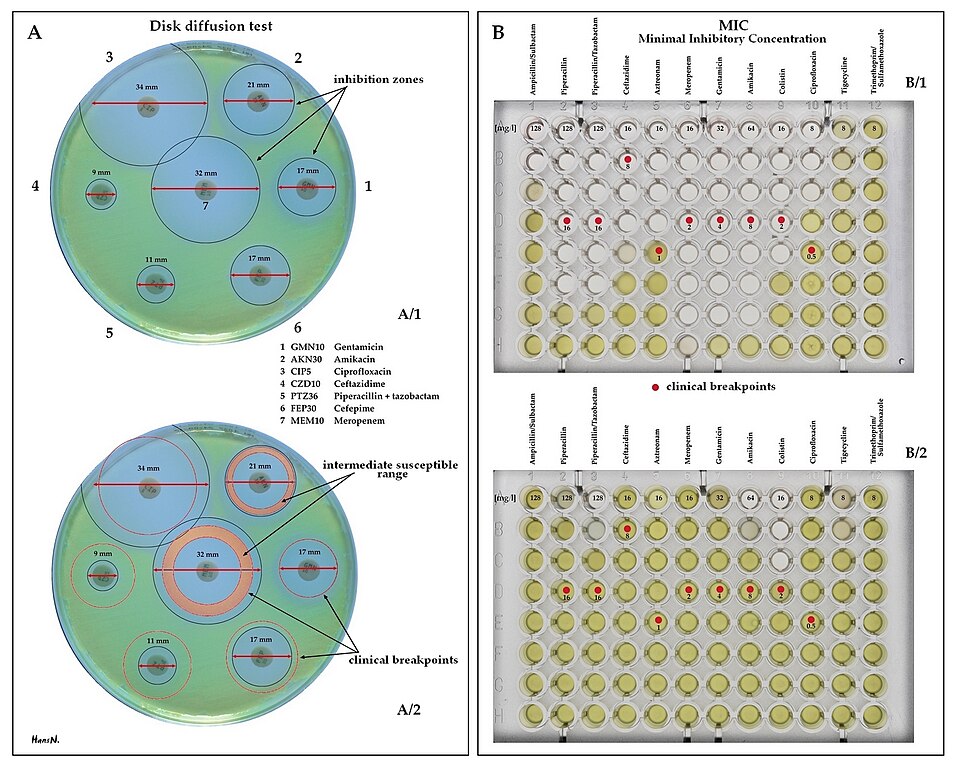
Overview
- Antibiotic resistance is natural selection operating in real time: bacteria with heritable mutations or acquired resistance genes survive antibiotic treatment, reproduce, and rapidly replace susceptible populations—a process directly observed in laboratories, hospitals, and the environment.
- Resistance evolves through multiple mechanisms including enzyme-mediated drug degradation, efflux pumps, target modification, and reduced membrane permeability, and spreads rapidly between unrelated bacterial species via horizontal gene transfer through plasmids, conjugation, transduction, and transformation.
- The World Health Organization designates antibiotic resistance a top global health threat, with drug-resistant infections estimated to have directly caused 1.27 million deaths in 2019, a toll projected to exceed 10 million per year by 2050 if current trajectories continue.
Antibiotic resistance stands as one of the most consequential demonstrations of evolution by natural selection that humanity has ever observed. Within years—sometimes months—of the clinical introduction of every major antibiotic drug, bacteria have evolved heritable mechanisms to survive it. The process is not metaphorical: bacteria carrying genetic variants that confer resistance to a given drug survive treatment while susceptible competitors die, reproduce rapidly in the cleared ecological space, and pass their resistance genes to offspring and lateral neighbors alike. The result, measured in hospitals on every continent, is a global public health emergency in which infections once reliably treatable by standard antibiotics are now killing patients for whom no effective therapy exists.4, 17
Discovery of antibiotics
The story of antibiotics begins on a September morning in 1928, when Alexander Fleming returned from holiday to his laboratory at St Mary's Hospital in London and noticed that a petri dish of Staphylococcus bacteria had been contaminated by a mold. The area of agar immediately surrounding the mold was clear: the bacterial colonies nearest the intruder had been dissolved. Fleming identified the mold as a species of Penicillium and published his findings the following year, reporting that its culture filtrate inhibited the growth of many Gram-positive bacteria while being relatively non-toxic to human leukocytes.1 He called the active substance penicillin, though he lacked the biochemical means to purify it in stable form and initially considered it most useful as an antiseptic for laboratory cultures.
It was not until the late 1930s that Howard Florey and Ernst Chain at the University of Oxford undertook the systematic purification of penicillin and demonstrated its therapeutic potential in animal and human trials. Their first clinical trial, conducted in 1941 on a British police officer severely ill with staphylococcal and streptococcal septicemia, showed dramatic improvement following penicillin injections—though the drug supply was exhausted before a cure could be completed.3 By 1944, mass production in the United States was supplying Allied military forces with enough penicillin to transform the treatment of wound infections. Fleming, Florey, and Chain shared the 1945 Nobel Prize in Physiology or Medicine for this work.3
In a June 1945 New York Times article, Fleming issued a prescient warning that antibiotic use in insufficient doses could allow bacteria to become resistant—a danger he had already observed in the laboratory—a caution he would repeat in his Nobel lecture later that year.2, 3 He was correct in a way even he may not have fully appreciated: penicillin-resistant strains of Staphylococcus aureus were detected in London hospitals as early as 1947, only six years after penicillin's first clinical use.4 The golden age of antibiotic discovery, spanning roughly the 1940s through the 1960s, produced more than a dozen major drug classes—aminoglycosides, tetracyclines, macrolides, cephalosporins, fluoroquinolones, and others—each representing a distinct mechanism of bacterial inhibition. And each, without exception, was followed within years to decades by the emergence of resistant strains.4, 5
How antibiotics work
Antibiotics are molecules that selectively target cellular processes essential to bacteria but absent from or significantly different in human cells, exploiting the evolutionary gulf between prokaryotic and eukaryotic cell biology (a divergence rooted in endosymbiosis). The major mechanistic classes target four primary processes. Cell wall synthesis inhibitors, including the beta-lactams (penicillins, cephalosporins, carbapenems) and glycopeptides (vancomycin), block the assembly of peptidoglycan, the rigid polymer that gives bacterial cell walls their structural integrity. Because human cells lack cell walls entirely, these drugs are inherently selective. Without a functional cell wall, bacteria cannot maintain osmotic pressure and lyse.5, 6
Protein synthesis inhibitors exploit the structural differences between bacterial 70S ribosomes and the eukaryotic 80S ribosome. Aminoglycosides and tetracyclines target the 30S ribosomal subunit, while macrolides, chloramphenicol, and lincosamides target the 50S subunit. Because of the substantial structural divergence between the prokaryotic and eukaryotic ribosomes, these antibiotics can selectively arrest bacterial protein production with limited effect on host cell ribosomes—though some toxicity arises because mitochondria, evolutionary descendants of proteobacteria, possess 70S ribosomes as well.5
DNA replication and transcription inhibitors form a third major class. Fluoroquinolones block bacterial DNA gyrase and topoisomerase IV, enzymes that relax supercoiled DNA ahead of the replication fork and that have no close functional equivalents in eukaryotic nuclei. Rifamycins inhibit bacterial RNA polymerase at a binding site not found in the mammalian enzyme. A fourth group—cell membrane disruptors such as polymyxins—targets the unique lipopolysaccharide outer membrane of Gram-negative bacteria, binding to lipid A and disrupting membrane integrity.5, 7
Mechanisms of resistance
Bacterial resistance to antibiotics arises through several distinct biochemical strategies, each of which represents an evolutionary solution to the selective pressure imposed by antibiotic exposure. The first and perhaps most thoroughly studied mechanism is enzymatic drug degradation. Beta-lactamase enzymes hydrolyze the beta-lactam ring that is the pharmacologically active component of penicillins, cephalosporins, and carbapenems, rendering the drug inactive before it can reach its target. Beta-lactamases are among the most diverse enzyme families known: by 2011, more than 1,000 distinct beta-lactamases had been characterized, organized into four molecular classes (A through D) based on their catalytic mechanism.6 Extended-spectrum beta-lactamases (ESBLs) hydrolyze third-generation cephalosporins that were developed specifically to escape earlier enzyme variants, while carbapenemases—including the KPC, NDM, and OXA families—inactivate the carbapenem drugs that were for decades the antibiotics of last resort against Gram-negative infections.6, 14
{kind=link}
A second major resistance mechanism involves efflux pumps: membrane-spanning protein complexes that actively export antibiotics from the bacterial cell before they can reach inhibitory concentrations. Bacteria possess several superfamilies of efflux pumps, including the RND (resistance-nodulation-cell division), MFS (major facilitator superfamily), and ABC transporter families. Many efflux pumps are broad-spectrum, capable of extruding structurally diverse antibiotics simultaneously—a phenomenon that contributes to multi-drug resistance. The MexAB-OprM efflux system of Pseudomonas aeruginosa, for example, exports beta-lactams, fluoroquinolones, tetracyclines, chloramphenicol, and other structurally unrelated compounds.7
Target modification is a third strategy. Rather than destroying the drug or preventing its intracellular accumulation, bacteria alter the molecular target so that the antibiotic can no longer bind it effectively. Methicillin resistance in Staphylococcus aureus is the classic example: the mecA gene encodes an alternative penicillin-binding protein (PBP2a) with low affinity for virtually all beta-lactam antibiotics, allowing cell wall synthesis to continue unimpaired in the presence of drugs that block the native PBPs.12 Fluoroquinolone resistance frequently arises from point mutations in the genes encoding DNA gyrase and topoisomerase IV that reduce the enzyme's affinity for the drug without significantly impairing its normal enzymatic function. Vancomycin resistance in enterococci involves a more elaborate reprogramming of cell wall biosynthesis: the VanA gene cluster encodes enzymes that replace the D-Ala-D-Ala terminus of peptidoglycan precursors—the site to which vancomycin binds—with D-Ala-D-Lac, which binds vancomycin approximately one thousand-fold less tightly.13
Reduced permeability constitutes a fourth mechanism. The outer membrane of Gram-negative bacteria contains protein channels called porins through which small hydrophilic molecules, including many antibiotics, must pass to enter the cell. Bacteria can downregulate or structurally modify specific porins, reducing drug uptake. Loss of the OmpF and OmpC porins in Escherichia coli, for instance, reduces susceptibility to beta-lactams, fluoroquinolones, and chloramphenicol.8 When porin loss is combined with upregulated efflux pump expression, the resulting reduction in intracellular antibiotic concentration can be orders of magnitude, conferring clinically significant resistance without any single mutation that would by itself be sufficient.7, 8
Horizontal gene transfer and the spread of resistance
What makes antibiotic resistance particularly dangerous from an evolutionary perspective is not merely that resistant mutants arise within a bacterial clone, but that resistance genes spread horizontally between unrelated bacterial lineages through processes collectively termed horizontal gene transfer (HGT). Unlike vertical gene transmission from parent to offspring, HGT allows a resistance mechanism that evolved in one species to be acquired almost immediately by entirely different species sharing the same environment—including species that have never been exposed to the selecting antibiotic.9
The primary vehicle for horizontal spread of antibiotic resistance is the plasmid: a circular, extrachromosomal DNA molecule capable of autonomous replication and, in many cases, self-transfer between bacterial cells through a process called conjugation. During conjugation, a donor cell forms a direct physical connection with a recipient cell and transfers a copy of the plasmid through a protein channel. Many resistance plasmids carry genes for multiple resistance mechanisms simultaneously—in some cases conferring resistance to five or more antibiotic classes on a single transferable element—allowing a single conjugation event to confer multi-drug resistance to a previously susceptible recipient.9, 10 Clinically, the spread of carbapenem resistance among Gram-negative bacteria has been driven largely by plasmids carrying carbapenemase genes such as blaKPC and blaNDM-1, which have disseminated globally across multiple bacterial genera including Klebsiella, Escherichia, Acinetobacter, and Pseudomonas.14
Bacteriophages—viruses that infect bacteria—can transfer bacterial DNA, including resistance genes, through a process called transduction. In specialized transduction, a phage accidentally packages a segment of chromosomal DNA flanking its integration site along with or instead of its own genome, and injects this DNA into a new bacterial host. In generalized transduction, phage capsids can be mistakenly packed with any segment of bacterial DNA during the packaging step of phage replication, enabling accidental transfer of any chromosomal or plasmid gene to a new cell.11 A third HGT mechanism, transformation, involves the uptake of free DNA from the environment by naturally competent bacteria—species capable of actively binding and internalizing extracellular DNA fragments released by lysed cells. Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria species are among the naturally competent pathogens in which transformation-mediated acquisition of resistance has been documented.9
Resistance genes also move within and between bacterial genomes via integrons and transposons—mobile genetic elements that can excise from one location and insert into another, often bringing associated resistance genes along. Class 1 integrons, in particular, are associated with a remarkably wide range of resistance gene cassettes and have been found in environmental bacteria, commensal bacteria, and clinical pathogens across multiple continents, suggesting a global pool of mobilizable resistance determinants that predates the antibiotic era.5, 9
The resistance timeline
The temporal pattern of resistance emergence relative to antibiotic introduction is one of the most striking features of the resistance crisis. Without exception, every antibiotic class introduced into clinical use has been followed by the detection of resistant strains, typically within a few years of widespread deployment. Penicillin G was in clinical use by 1942 and penicillinase-producing staphylococci were documented in 1947—a gap of five years.4 Methicillin was developed to overcome penicillinase and introduced in 1959; MRSA was identified in 1961, a gap of two years.12 Vancomycin, considered a drug of last resort for Gram-positive infections, was approved in 1958; vancomycin-resistant enterococci (VRE) were first reported in Europe in 1986 and 1988, a gap of 28 to 30 years—the longest interval on record, though notably vancomycin use was relatively restricted during much of that period.13
Antibiotic introduction and first reported clinical resistance4, 5, 12, 13, 14
| Antibiotic | Class | Clinical introduction | Resistance first reported | Interval (years) |
|---|---|---|---|---|
| Penicillin G | Beta-lactam | 1942 | 1947 | 5 |
| Streptomycin | Aminoglycoside | 1943 | 1947 | 4 |
| Chloramphenicol | Phenicol | 1948 | 1950 | 2 |
| Tetracycline | Tetracycline | 1952 | 1956 | 4 |
| Erythromycin | Macrolide | 1952 | 1955 | 3 |
| Methicillin | Beta-lactam | 1959 | 1961 | 2 |
| Ampicillin | Beta-lactam | 1961 | 1973 | 12 |
| Cephalosporins (1st gen.) | Beta-lactam | 1964 | 1966 | 2 |
| Vancomycin | Glycopeptide | 1958 | 1988 | 30 |
| Imipenem (carbapenem) | Beta-lactam | 1985 | 1998 | 13 |
| Linezolid | Oxazolidinone | 2000 | 2001 | 1 |
| Daptomycin | Lipopeptide | 2003 | 2005 | 2 |
Some scholars have noted that resistance determinants against antibiotics predating clinical use by decades have been found in ancient permafrost DNA and in the microbiomes of isolated human populations with no history of antibiotic exposure, demonstrating that resistance genes are part of the natural evolutionary history of bacteria and were not created by the antibiotic era.5 The clinical antibiotic era did not invent resistance; it applied a powerful selection pressure that dramatically increased the frequency and clinical significance of resistance mechanisms that already existed in the microbial world at low prevalence.5, 9
Superbugs and the global crisis
The term "superbug" is colloquially applied to bacterial strains that are resistant to multiple antibiotic classes simultaneously. The most clinically significant include MRSA, vancomycin-resistant enterococci (VRE), carbapenem-resistant Enterobacteriaceae (CRE), and extensively drug-resistant tuberculosis (XDR-TB). The WHO published its first global priority pathogen list in 2017, ranking 12 bacterial families by urgency of new therapeutic need. The critical-priority group—the highest tier—comprised carbapenem-resistant Acinetobacter baumannii, carbapenem-resistant Pseudomonas aeruginosa, and carbapenem- or third-generation-cephalosporin-resistant Enterobacteriaceae.16
_Bacteria.jpg)
_Bacteria.jpg){kind=link}
MRSA has been particularly well-studied. Phylogenomic analyses have traced the origin of the hospital-associated MRSA lineages to the acquisition of the mecA gene on the staphylococcal cassette chromosome mec (SCCmec) from a related coagulase-negative staphylococcal species, most probably Staphylococcus sciuri or a related taxon.12 MRSA diversified from a small number of ancestral clones into the distinct healthcare-associated (HA-MRSA) and community-associated (CA-MRSA) lineages that now circulate globally. CA-MRSA strains, which emerged in the 1990s outside hospital settings, frequently carry Panton-Valentine leukocidin toxin genes and cause severe skin and soft tissue infections, pneumonia, and bloodstream infections in otherwise healthy young people.12
Tuberculosis, caused by Mycobacterium tuberculosis, presents a distinct and particularly grave resistance problem. Multi-drug-resistant tuberculosis (MDR-TB) is defined as resistance to both isoniazid and rifampicin, the two most effective first-line agents. XDR-TB additionally involves resistance to any fluoroquinolone and at least one of the injectable second-line drugs (amikacin, kanamycin, or capreomycin). The WHO estimated 410,000 incident cases of rifampicin-resistant TB in 2022, of which approximately 63% were MDR-TB, with an overall treatment success rate for MDR-TB of only 63%.15 Pre-XDR and XDR strains represent a still more severe clinical situation, with treatment success rates frequently below 40% and treatment courses lasting 18 to 24 months or more.15
A comprehensive 2022 analysis in The Lancet, drawing on data from 204 countries and territories, estimated that bacterial antimicrobial resistance was directly responsible for 1.27 million deaths globally in 2019, with an additional 4.95 million deaths in which resistance contributed as an associated factor. Lower respiratory infections, bloodstream infections, and intra-abdominal infections accounted for the largest shares of the attributable mortality burden.17 The Review on Antimicrobial Resistance commissioned by the UK government projected that, on current trajectories, drug-resistant infections could kill 10 million people per year by 2050—more than current deaths from cancer—with cumulative economic losses of $100 trillion.18
Direct deaths attributable to antimicrobial resistance by infectious syndrome, 201917
Antibiotic resistance as evolution made visible
The evolutionary dynamics of antibiotic resistance have been studied in remarkable experimental detail, producing some of the most visually and conceptually compelling demonstrations of natural selection on record. In 2016, Michael Baym and colleagues at Harvard Medical School constructed what they called the "MEGA-plate" experiment—a large (120 cm × 60 cm) petri dish divided into lanes, each containing progressively higher concentrations of the antibiotic trimethoprim from the edges to the center. A population of Escherichia coli was inoculated at the edges and filmed continuously as it evolved. The bacteria advanced through the first antibiotic zone in hours, evolved resistance to the next concentration within a day or two, and ultimately crossed the entire plate—developing resistance to a concentration 1,000 times the initial minimum inhibitory concentration—in approximately 11 days.19
_Bacteria_colorized_scanning_electron_micrograph_-_44.jpg){kind=link}
The MEGA-plate experiment made visibly apparent several principles that had been established by earlier work in less dramatic form. Resistance did not emerge simultaneously across the expanding front; it evolved repeatedly and independently in different subpopulations, with the most resistant mutants arising at the boundaries where selection pressure was highest. Once a highly resistant clone appeared, it rapidly expanded into the higher-concentration zone, leaving behind a trail of intermediate-resistance cells whose descendants then continued to evolve toward still-higher concentrations. The experiment illustrated that evolution under antibiotic selection is not a single-step event but a stepwise process in which each resistant mutant provides a platform for the evolution of still greater resistance—precisely the mechanism of incremental natural selection.19
This experimental observation is complemented by Richard Lenski's long-term evolution experiment (LTEE), begun in 1988 with twelve replicate populations of E. coli propagated in identical glucose-limited media. After more than 75,000 generations, all populations have continued to improve in fitness relative to their ancestor, demonstrating that adaptation under laboratory conditions is not bounded in any short timescale.20, 21 The LTEE populations show both parallel evolution—the same mutations arising independently in multiple lineages—and historical contingency—unique evolutionary trajectories dependent on the specific sequence of mutations that accumulated in each population. Both patterns are observed in clinical antibiotic resistance: resistance mutations in different patients and different hospitals often converge on the same molecular mechanisms, yet each strain carries a unique complement of additional mutations that influence fitness, transmission, and virulence.20
Agricultural overuse and environmental reservoirs
Roughly half of all antibiotics produced globally are used not to treat human infections but to promote growth and prevent disease in food-producing animals, particularly in industrial livestock and aquaculture operations.22 In many countries, the mass of antibiotics administered to animals substantially exceeds that used in human medicine; in the United States prior to regulatory reforms in 2017, approximately 70% of medically important antibiotics sold were for use in food animals.23 Sub-therapeutic antibiotic doses in animal agriculture create precisely the conditions most favorable for the evolution of resistance: sustained exposure to antibiotic concentrations that kill susceptible bacteria but allow resistant variants to survive and proliferate.22, 23
Resistance genes and resistant bacteria move from agricultural settings into the environment through animal waste, which contaminates soil and waterways, and through the food supply itself, where resistant bacteria on meat and produce can colonize human intestinal tracts. The colistin-resistance gene mcr-1, a transferable plasmid-borne gene that appeared in Chinese livestock in 2015 and rapidly spread to human clinical isolates and environmental bacteria on multiple continents, is perhaps the most alarming recent example of this pathway.23 Colistin had been considered a last-line drug for carbapenem-resistant Gram-negative infections; the emergence and rapid global spread of plasmid-mediated colistin resistance eliminated it as a reliable therapeutic option for the most difficult-to-treat infections in many clinical settings.16, 23
The environment more broadly functions as a vast reservoir of resistance genes. Soil bacteria, many of which are naturally antibiotic-producing organisms, have maintained arsenals of resistance mechanisms for millions of years as part of their ecological interactions with antibiotic-producing competitors. This natural reservoir, combined with the selective amplification of resistance caused by clinical and agricultural antibiotic use, and the remarkable mobility of resistance genes through horizontal transfer, creates a resistance gene pool far larger and more dynamic than any clinical or agricultural setting alone would generate.5
Combating resistance
Responses to the antibiotic resistance crisis operate at multiple levels: reducing unnecessary antibiotic use through stewardship programs, developing new drugs and therapeutic alternatives, and coordinating global surveillance and policy. Antibiotic stewardship programs in hospitals aim to ensure that antibiotics are prescribed only when indicated, that the narrowest-spectrum effective drug is chosen, and that treatment durations are minimized, all of which reduce the selective pressure driving resistance evolution. A systematic review of hospital stewardship programs found that they reduce antibiotic consumption by 20 to 35% without adverse effects on patient outcomes.27
The antibiotic discovery pipeline has been in crisis since approximately 1987, when the last truly novel antibiotic class—the lipopeptides, exemplified by daptomycin—was discovered, creating what researchers call the "discovery void."4 The pharmaceutical industry largely exited antibiotic development in the 1990s and 2000s, deterred by the economics of infectious disease drugs (which are used for short courses rather than chronically, limiting profitability) and by the scientific difficulty of finding new mechanisms of action in a target landscape well-explored by decades of discovery chemistry. The 2015 announcement of teixobactin, discovered using a new cultivation technique for unculturable soil bacteria, provided a rare example of a genuinely new antibiotic class active against Gram-positive bacteria, with low initial resistance rates attributed to its mechanism of binding lipid II precursors at multiple sites rather than a single protein target.26
Artificial intelligence is increasingly applied to antibiotic discovery. A 2024 study in Nature by Wong and colleagues described a deep learning approach that identified a structural class of antibiotics from a molecular library, with the model trained on bacterial growth inhibition data and used to search chemical space for novel structural scaffolds. Candidate compounds identified by the model showed activity against several drug-resistant pathogens including MRSA and vancomycin-resistant Enterococcus faecalis in mouse infection models.25 Though such approaches remain early-stage, they represent a qualitative expansion of the chemical space accessible to antibiotic discovery programs beyond the natural product chemistry that dominated the golden age of discovery.25
Bacteriophage therapy—the therapeutic use of bacteriophages, viruses that selectively infect and kill bacteria—has attracted renewed interest as an alternative or complement to conventional antibiotics.24 Phage therapy was widely used in Eastern Europe throughout the twentieth century but fell out of favor in the West following the success of antibiotics. Modern phage therapy programs, typically involving genetically characterized and purified phage preparations, have demonstrated efficacy in case reports and small series against infections with drug-resistant organisms including MRSA and carbapenem-resistant Acinetobacter that had failed all conventional treatments.24 The evolutionary dynamics of phage-bacteria interactions are themselves a subject of research: bacteria can evolve resistance to phages, but phages can in turn evolve to counter bacterial resistance, suggesting that phage therapy may require ongoing evolutionary management analogous to the principles of antibiotic stewardship.24
The WHO Global Action Plan on Antimicrobial Resistance, endorsed by all member states in 2015, calls for surveillance of resistance patterns, reduction of antimicrobial use in human medicine and agriculture, and investment in new drug development.27 The Lancet analysis estimated that increasing access to existing antibiotics in low-income countries, where under-treatment of susceptible infections remains a major cause of death, could prevent as many resistance-related deaths as restricting overuse in high-income settings, underscoring that the global response must address both overuse and underuse simultaneously.17
Evolutionary significance
Antibiotic resistance is frequently cited in evolutionary biology as the clearest and most consequential real-time demonstration of natural selection, because it fulfills every requirement of the Darwinian mechanism—heritable variation, differential survival, and change in population composition over time (see mutation and DNA repair)—in a system directly observable with standard microbiological tools.4, 5 The MEGA-plate experiment made visible in a single photograph what Darwin could only infer from geological and paleontological evidence: populations evolving under directional selection change incrementally, with each successful variant building on the last.19
The spread of resistance through horizontal gene transfer illuminates a dimension of evolution that Darwin did not anticipate but that is now recognized as a major force in bacterial evolution: the transfer of genetic information between lineages rather than only from parent to offspring. The global dissemination of carbapenemase genes, traveling in hours on plasmids between patients in different countries via the movement of colonized individuals, is an evolutionary event of extraordinary speed—far faster than any process driven by mutation alone.9, 14
The resistance crisis also demonstrates with painful clarity the practical consequences of ignoring evolutionary principles in medicine and agriculture. Every clinical decision to prescribe an antibiotic unnecessarily, every agricultural practice that exposes billions of bacteria to sub-lethal antibiotic concentrations, is an act of artificial selection for resistance. The bacteria do not choose to evolve; selection acts on pre-existing variation and produces adaptation as an inevitable outcome. Understanding this process at a mechanistic level is not an academic exercise. It is the foundation on which rational antibiotic use, drug development strategy, and global health policy must be built.4, 17, 27
References
On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzae
Antibiotic resistance mechanisms in bacteria: relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens
Beta-lactamases in the 21st century: characterization, epidemiology and detection of this important resistance threat
Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics
Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations
Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli
Antibiotic use in food-producing animals: overview of the current regulatory situation in Europe
Teixobactin, the first member of a new class of antibiotics, kills bacteria without detectable resistance