Overview
- Genetic drift is the random fluctuation of allele frequencies in finite populations due to chance sampling in reproduction, and its effects are strongest in small populations where stochastic variation can override the deterministic force of natural selection.
- The neutral theory of molecular evolution, proposed by Motoo Kimura in 1968, demonstrated that most evolutionary change at the molecular level is driven by the random fixation of selectively neutral mutations rather than by adaptive natural selection.
- Population bottlenecks and founder effects have shaped the genetic architecture of numerous species, including humans, by drastically reducing genetic diversity and elevating the frequency of otherwise rare alleles through chance alone.
Genetic drift is the change in allele frequency in a population due to random sampling of organisms in each generation. Unlike natural selection, which favours alleles that increase an organism's fitness, genetic drift is entirely stochastic: alleles may increase or decrease in frequency by chance alone, regardless of whether they are beneficial, neutral, or harmful. Drift is one of the four fundamental mechanisms of evolutionary change, alongside natural selection, mutation, and gene flow, and its effects are most pronounced in small populations where random fluctuations in allele frequency are not averaged out over large numbers of individuals.1, 5 First described mathematically by the American geneticist Sewall Wright in the 1930s, genetic drift has since been recognised as a central force in shaping the genetic composition of populations, and its theoretical elaboration into the neutral theory of molecular evolution by Motoo Kimura in 1968 transformed the understanding of how genomes change over time.1, 2
The mechanism of drift
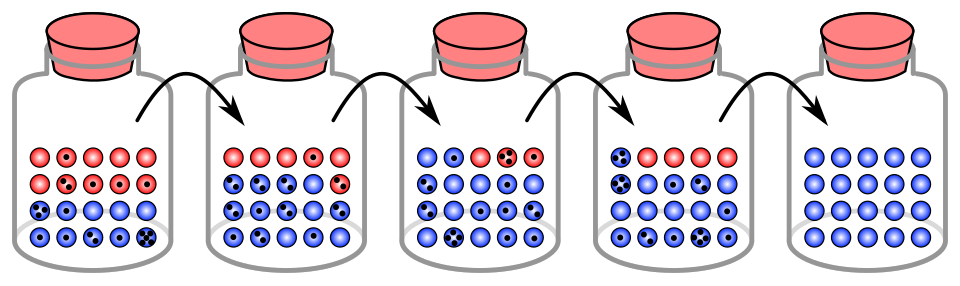
In any sexually reproducing population, the alleles transmitted from one generation to the next represent a sample of the alleles present in the parental generation.
{kind=link}
If the population is finite, as all real populations are, this sampling process introduces random variation into allele frequencies that is analogous to the statistical noise inherent in drawing a limited number of items from a larger pool. Consider a population in which a particular allele exists at a frequency of 50 percent. If the population consists of thousands of individuals, the allele frequency in the next generation will almost certainly remain very close to 50 percent, because the law of large numbers ensures that the sample closely approximates the true frequency. If, however, the population consists of only ten individuals, the allele frequency may fluctuate substantially from one generation to the next simply because each individual's contribution to the next generation is subject to chance variation in mating success, fecundity, and survival.5, 18
The process is often compared to coin flipping. A fair coin has an equal probability of landing heads or tails on any single flip, but a sequence of ten flips will frequently produce ratios of seven-to-three or even eight-to-two by chance alone, whereas a sequence of ten thousand flips will almost invariably yield a ratio very close to fifty-fifty. In the same way, genetic drift produces larger fluctuations in smaller populations, and these fluctuations accumulate over generations.18 Over sufficient time, drift will inevitably drive every allele in a finite population to one of two absorbing states: fixation, in which the allele reaches a frequency of 100 percent and becomes the only variant at that locus, or loss, in which the allele disappears entirely. For a new neutral mutation arising in a diploid population of effective size Ne, the probability of eventual fixation is simply 1/(2Ne), because the mutation initially exists as a single copy among 2Ne total alleles at that locus.2, 5
The rate at which allele frequencies fluctuate due to drift is inversely proportional to the population size. Wright showed that the variance in allele frequency change per generation for a neutral allele at frequency p is approximately p(1 − p)/(2Ne), where Ne is the effective population size.1 This means that drift operates more rapidly in small populations: alleles reach fixation or loss more quickly, and genetic diversity is eroded at a faster pace. In very large populations, the per-generation change in allele frequency due to drift is negligible, and the deterministic force of natural selection dominates the evolutionary trajectory of most alleles.1, 6
The most celebrated experimental demonstration of genetic drift was performed by Peter Buri in 1956. Buri established 107 replicate populations of Drosophila melanogaster, each consisting of exactly 16 individuals heterozygous for two alleles of the brown eye-colour gene (bw and bw75), so that both alleles began at a frequency of 50 percent in every population. The populations were maintained at constant size for 19 generations with no selection acting on the eye-colour locus. By generation 19, the bw75 allele had been lost entirely from 30 populations and had drifted to fixation in 28 others, while the remaining populations showed allele frequencies scattered across the full range from near-zero to near-one. The distribution of allele frequencies across the 107 populations closely matched the theoretical predictions of the Wright-Fisher model, providing a textbook confirmation that random sampling alone, in the absence of selection, is sufficient to drive alleles to fixation or loss in small populations.24, 18
Effective population size
The concept of effective population size (Ne) is central to the theory of genetic drift. Introduced by Wright in 1931, Ne is defined as the size of an idealised population that would experience the same rate of genetic drift as the actual population under consideration.1 In an idealised Wright-Fisher population, all individuals are diploid, mating is random, generations are non-overlapping, and every individual has an equal probability of contributing gametes to the next generation. Real populations violate these assumptions in numerous ways, and as a result, the effective population size is almost always substantially smaller than the census population size (N).6
Several factors reduce Ne relative to N. Unequal sex ratios concentrate reproduction in fewer individuals: if a population of 100 consists of 10 males and 90 females, the effective population size is not 100 but approximately 36, because the rarer sex disproportionately constrains the genetic sampling process.5, 18 High variance in reproductive success has a similar effect, because a population in which a few individuals produce most of the offspring behaves genetically as though it were much smaller than its census count. Fluctuations in population size over time are particularly important: the effective population size over multiple generations is determined by the harmonic mean of the population sizes in each generation, which is dominated by the smallest values. A population that alternates between 10,000 and 100 individuals has a long-term effective size closer to 200 than to 5,050.6, 18
The practical consequences of these reductions are striking. Humans have a current census population of approximately eight billion, but estimates of the long-term effective population size of the human lineage are remarkably small, ranging from roughly 10,000 to 15,000 individuals. This figure reflects the cumulative impact of ancestral population bottlenecks, particularly during the out-of-Africa migration approximately 50,000 to 70,000 years ago, and the generally small population sizes that characterised most of human prehistory.15, 9 The effective population size determines the boundary between selection-dominated and drift-dominated evolutionary regimes: when the product of Ne and the selection coefficient s is much greater than one, selection efficiently governs the fate of an allele, but when Nes is much less than one, the allele's trajectory is determined primarily by drift.6, 7
The founder effect and population bottlenecks

{kind=link}
Two special cases of genetic drift have particularly dramatic consequences for the genetic composition of populations: the founder effect and the population bottleneck. In the founder effect, a small number of individuals from a larger population establish a new colony in a geographically isolated area. Because the founders carry only a subset of the genetic variation present in the source population, the new population begins with allele frequencies that may differ substantially from those of the parent group. Rare alleles in the source population may be present at high frequency among the founders, or conversely, common alleles may be absent entirely.4, 18
Some of the most striking examples of the founder effect involve human populations. The Afrikaner population of South Africa traces much of its ancestry to a small number of Dutch settlers who arrived at the Cape of Good Hope in the seventeenth century. Among these founders was an individual who carried a mutation in the protoporphyrinogen oxidase gene (the R59W variant) that causes variegate porphyria, a disorder of haem biosynthesis. Because the founding population was small and the community remained relatively insular, this mutation rose to a frequency far higher among Afrikaners than in any other population worldwide, with approximately 95 percent of South African variegate porphyria cases carrying this single founder mutation.19 Similarly, the Old Order Amish of Lancaster County, Pennsylvania, descend from a small group of eighteenth-century immigrants, and among them the recessive allele for Ellis-van Creveld syndrome, a form of short-limbed dwarfism, occurs at an unusually high frequency traceable to the original founders.23
On the remote Pacific atoll of Pingelap in Micronesia, a devastating typhoon in approximately 1775 reduced the population to an estimated 20 survivors. Among these survivors, at least one individual carried a recessive mutation in the CNGB3 gene that causes achromatopsia, or complete colour blindness. In the centuries since, the population has recovered to several hundred individuals, but the frequency of achromatopsia on Pingelap is between 4 and 10 percent, roughly a thousandfold higher than in most other populations, a direct consequence of the genetic bottleneck imposed by the typhoon.20
A population bottleneck occurs when a population undergoes a drastic reduction in size, regardless of whether a new colony is being established. The genetic consequences are similar to those of the founder effect: allelic diversity is lost, and the allele frequencies in the post-bottleneck population may differ markedly from those in the pre-bottleneck population. Nei, Maruyama, and Chakraborty showed mathematically that a bottleneck rapidly reduces heterozygosity and the number of alleles in a population, and that while heterozygosity can recover relatively quickly through new mutations once the population expands, the full spectrum of allelic diversity takes much longer to be restored.4
Notable population bottlenecks and founder effects4, 10, 11, 19, 20, 23
| Population | Type | Bottleneck size | Genetic consequence |
|---|---|---|---|
| Northern elephant seal | Bottleneck | ~20 individuals (1890s) | Extremely low genetic variation despite recovery to >100,000 |
| Cheetah (Acinonyx jubatus) | Bottleneck | Unknown (Late Pleistocene) | Near-total genetic uniformity; reciprocal skin grafts accepted |
| Afrikaner population | Founder effect | ~1,000 settlers (1600s) | High frequency of variegate porphyria (R59W mutation) |
| Old Order Amish | Founder effect | ~200 immigrants (1700s) | Elevated Ellis-van Creveld syndrome prevalence |
| Pingelap atoll | Bottleneck | ~20 survivors (c. 1775) | 4–10% prevalence of achromatopsia |
| Ashkenazi Jewish population | Founder effect | ~350–500 founders (medieval) | Elevated frequency of multiple recessive disease alleles |

{kind=link}
The northern elephant seal provides one of the most dramatic examples of a population bottleneck in a non-human species. By the late nineteenth century, commercial hunting had reduced the population of Mirounga angustirostris to as few as 20 individuals. Under legal protection, the population has since recovered to more than 100,000 animals, but genetic surveys reveal strikingly low levels of variation: Hoelzel and colleagues found only two mitochondrial DNA control region haplotypes in the northern species, compared with 23 haplotypes in the closely related southern elephant seal, which did not experience a comparable bottleneck.10 The cheetah presents a similar pattern. O'Brien and colleagues demonstrated in 1983 that cheetahs are so genetically uniform that reciprocal skin grafts between unrelated individuals are accepted without immune rejection, a finding consistent with a severe population bottleneck during the late Pleistocene that reduced genetic diversity to near homogeneity.11
The neutral theory of molecular evolution
The neutral theory of molecular evolution, proposed by Motoo Kimura in 1968, fundamentally reoriented the understanding of genetic drift from a minor evolutionary nuisance to the predominant driver of molecular evolutionary change. Kimura observed that the rate of amino acid substitution inferred from comparisons of protein sequences across species was far higher than could be accounted for by positive natural selection, because the required intensity of selection would impose an intolerable genetic load on the population.2 He proposed instead that the vast majority of mutations that become fixed in populations are selectively neutral: they have no appreciable effect on the fitness of the organism, and their fate is determined entirely by genetic drift.2, 7
A central prediction of the neutral theory is elegant in its simplicity. If the rate at which new neutral mutations arise in a diploid population is 2Neμ per generation (where μ is the per-individual mutation rate), and the probability that any given neutral mutation ultimately reaches fixation is 1/(2Ne), then the rate of neutral substitution is the product of these two quantities: 2Neμ × 1/(2Ne) = μ. The rate of neutral molecular evolution is therefore equal to the mutation rate and is independent of population size.2, 7 This result provides the theoretical foundation for the molecular clock, the observation that neutral DNA sequences accumulate substitutions at an approximately constant rate per unit time, which has become one of the most widely used tools in molecular phylogenetics for estimating divergence times between lineages.7
Kimura elaborated the neutral theory in a comprehensive monograph in 1983, marshalling evidence from protein electrophoresis, DNA sequence comparisons, and population-genetic theory to argue that neutral drift, not adaptive selection, accounts for most of the molecular variation observed both within and between species.7 The theory does not deny the importance of natural selection at the phenotypic level; rather, it asserts that at the level of individual nucleotide and amino acid substitutions, the majority of changes are selectively inconsequential and evolve by drift. This distinction between molecular and phenotypic evolution was initially controversial but has become a foundational principle of modern evolutionary genetics.7, 17
The nearly neutral theory
In 1973, Tomoko Ohta extended the neutral theory with her nearly neutral theory, which incorporated mutations whose selective effects are not strictly zero but are so small that their fate is governed largely by drift rather than by selection. Ohta proposed that a substantial fraction of molecular mutations are slightly deleterious rather than perfectly neutral, and that whether these mutations behave as effectively neutral depends on the effective population size of the organism in question.3
The critical threshold is determined by the relationship between the effective population size Ne and the selection coefficient s of the mutation. When the absolute value of Nes is much less than one, the selective effect of the mutation is too weak to overcome the stochastic noise of drift, and the mutation behaves as if it were neutral. When Nes is much greater than one, selection efficiently determines the mutation's fate. The nearly neutral theory therefore predicts that small populations will accumulate slightly deleterious mutations at a higher rate than large populations, because mutations that would be eliminated by selection in a large population can drift to fixation in a small one.3, 6
This prediction has been confirmed by comparative genomic studies. Species with small effective population sizes tend to exhibit higher ratios of nonsynonymous to synonymous substitutions (dN/dS ratios), indicating that a greater proportion of amino acid changes have reached fixation in these lineages, consistent with the fixation of slightly deleterious mutations by drift.6, 21 The nearly neutral theory also explains why the molecular clock is not perfectly constant: lineages with smaller effective population sizes are expected to have slightly faster rates of molecular evolution at functionally constrained sites, because the boundary between neutral and selected mutations shifts upward as Ne decreases.3, 7
Drift versus selection
The relative importance of genetic drift and natural selection in shaping evolutionary change depends on the product Nes, which measures the strength of selection relative to the intensity of drift. When Nes is much greater than one, selection is the dominant force: beneficial mutations are efficiently fixed, and deleterious mutations are efficiently purged. When Nes is much less than one, drift overpowers selection, and the fate of alleles is determined by chance. This relationship means that the same mutation can be visible to selection in a large population but effectively invisible in a small one.6, 8
The practical consequences of this interplay are profound. In small populations, slightly beneficial mutations may be lost by drift before selection can drive them to fixation, reducing the rate of adaptive evolution. Conversely, slightly deleterious mutations that would be eliminated by purifying selection in a large population may drift to fixation in a small one, gradually degrading the functional integrity of the genome over time.6, 22 This irreversible accumulation of deleterious mutations in small asexual populations was described by Hermann Muller in 1964 as Muller's ratchet: because an asexual population cannot regenerate mutation-free genotypes through recombination, the least-loaded class of individuals is lost to drift with a probability that increases as the population shrinks, and each loss clicks the ratchet one notch further toward genomic deterioration.13
The tension between drift and selection is not merely of academic interest. Endangered species with small population sizes face a dual genetic threat: the immediate loss of adaptive genetic variation through drift, which reduces the population's ability to respond to environmental challenges, and the gradual accumulation of mildly deleterious mutations, which depresses the mean fitness of the population over time. Frankham reviewed the evidence for a causal link between low genetic diversity and elevated extinction risk and concluded that genetic factors, including inbreeding depression and the fixation of deleterious alleles by drift, contribute materially to the vulnerability of small populations.16 This genetic erosion can create a feedback loop in which declining fitness reduces population size, which accelerates further genetic deterioration, a process sometimes called an extinction vortex.16
Drift in human evolution
Genetic drift has played a substantial role in shaping the genetic architecture of modern human populations. The out-of-Africa migration, during which a subset of the African population expanded into Eurasia and eventually colonised every major landmass, imposed a series of founder effects that progressively reduced genetic diversity with increasing distance from East Africa. Ramachandran and colleagues demonstrated in 2005 that a strong linear relationship exists between the geographic distance of a population from sub-Saharan Africa (measured along plausible migration routes) and its level of neutral genetic diversity, as measured by microsatellite heterozygosity. This pattern, termed the serial founder effect model, indicates that each successive colonisation event involved a small founding group that carried only a fraction of the diversity present in the source population.9
Prugnolle, Manica, and Balloux independently confirmed this geographic gradient in 2005, showing that distance from East Africa is the single best predictor of neutral genetic diversity across global human populations, explaining a majority of the observed variance in heterozygosity.14 Populations in sub-Saharan Africa harbour the greatest genetic diversity of any human group, while populations at the geographic extremes of the human range, such as indigenous peoples of South America and Oceania, tend to have the lowest diversity. This gradient is not a consequence of differential natural selection across environments but rather a straightforward prediction of the serial founder effect model, in which each bottleneck at the wavefront of expansion randomly eliminates a portion of the genetic variation carried forward.9, 14, 15
The elevated frequency of certain disease-associated alleles in specific human populations provides further evidence of drift's influence. In the Ashkenazi Jewish population, which experienced a well-documented population contraction during the medieval period, several recessive disease alleles occur at frequencies far higher than in other populations. Risch and colleagues analysed the geographic distribution of disease mutations among Ashkenazi Jews and found that the pattern was more consistent with founder effects and genetic drift than with heterozygote advantage driven by natural selection, concluding that chance sampling during population bottlenecks is the most parsimonious explanation for the elevated carrier frequencies of conditions such as Tay-Sachs disease, Gaucher disease, and familial dysautonomia.12
The human effective population size during much of prehistory was far smaller than the current census count, and this history of small Ne has left a detectable imprint on the human genome. Lynch has argued that the relatively small long-term effective population size of humans, combined with the species' high per-generation mutation rate, results in a weaker efficiency of purifying selection against mildly deleterious mutations compared with organisms that maintain larger effective population sizes. This observation has implications for the long-term accumulation of genetic load in human populations and for understanding why the human genome contains a relatively high proportion of mildly deleterious variants compared with species such as Drosophila or bacteria that maintain effective population sizes orders of magnitude larger.22, 21
Detecting and measuring drift
Distinguishing the genomic signatures of drift from those of natural selection is a central challenge of population genetics. Under the neutral theory, drift produces predictable patterns of variation: allele frequencies across loci should conform to the expectations of the Wright-Fisher diffusion model, the site frequency spectrum (the distribution of allele frequencies across segregating sites) should follow a characteristic shape, and levels of divergence between species should correlate with levels of polymorphism within species, since both are driven by the same neutral mutation rate.7, 17
Deviations from these neutral expectations provide evidence for the action of selection. An excess of high-frequency derived alleles, for instance, can indicate a recent selective sweep in which a beneficial mutation hitchhiked linked neutral variants to high frequency, as captured by the H statistic of Fay and Wu.17 Conversely, an excess of rare variants compared with the neutral expectation may indicate purifying selection removing deleterious alleles, or a recent population expansion that has introduced many new mutations at low frequency. The challenge lies in the fact that demographic history, including bottlenecks, expansions, and population structure, can produce patterns in the site frequency spectrum that mimic the signatures of selection. Disentangling these effects requires either explicit demographic modelling or the use of methods that contrast patterns across different functional categories of the genome, such as comparisons between coding and non-coding regions or between replacement and synonymous sites.6, 17
Genome-wide surveys of variation have increasingly confirmed the predictions of the neutral and nearly neutral theories. The majority of nucleotide sites that differ between closely related species are in non-coding or synonymous positions where changes are unlikely to affect protein function, consistent with the expectation that most fixed differences are neutral.7 At the same time, the fraction of amino acid substitutions driven by positive selection varies across lineages and functional categories, and identifying the relative contributions of drift and selection to the evolution of any given gene or genomic region remains an active and technically demanding area of research.6, 17
Significance for evolutionary biology
The recognition of genetic drift as a major evolutionary force has had far-reaching consequences for biology. Before Wright's theoretical work in the 1930s and Kimura's neutral theory in 1968, the prevailing view was that virtually all evolutionary change was driven by natural selection, a position sometimes called panselectionism. The demonstration that drift is a powerful force in its own right, and that the majority of molecular evolutionary changes may be selectively neutral, introduced a null hypothesis against which the action of selection must be tested.1, 2, 7 This conceptual shift transformed molecular evolution into a quantitative, hypothesis-testing science rather than a discipline dominated by adaptive storytelling.
Drift also provides the theoretical foundation for the molecular clock, which enables the estimation of divergence times between lineages from DNA sequence data and underpins much of modern molecular phylogenetics.7 In conservation biology, the theory of drift informs the management of endangered species by establishing minimum viable population sizes necessary to maintain genetic diversity, by predicting the rate at which small populations will lose adaptive potential, and by guiding strategies for genetic rescue through the introduction of individuals from genetically distinct populations.16
The interplay between drift and selection remains one of the most actively investigated questions in evolutionary genetics. Advances in whole-genome sequencing have made it possible to characterise the full spectrum of variation within and between populations, to estimate effective population sizes with increasing precision, and to quantify the proportion of molecular evolution attributable to drift versus adaptation across diverse taxa. What is now clear is that evolution is not a simple story of organisms being sculpted by natural selection alone: chance, embodied in the stochastic sampling of alleles from generation to generation, is an inescapable and pervasive force in the history of life.6, 7, 8
References
Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa
Elephant seal genetic variation and the use of simulation models to investigate historical population bottlenecks
Geographic distribution of disease mutations in the Ashkenazi Jewish population supports genetic drift over selection
A R59W mutation in human protoporphyrinogen oxidase results in decreased enzyme activity and is prevalent in South Africans with variegate porphyria