Overview
- Genetic load is the reduction in mean fitness of a population relative to the fitness of the optimal genotype, a concept formalised by J. B. S. Haldane in the late 1930s that quantifies the cumulative cost of maintaining deleterious alleles through mutation, segregation, drift, and incomplete adaptation to changing environments.
- Mutation load arises from the continuous influx of deleterious mutations and, under mutation-selection balance, equals approximately the genomic mutation rate regardless of individual mutation effect sizes — a result known as the Haldane-Muller principle that implies even modest per-locus rates generate substantial aggregate load when spread across thousands of genes.
- Empirical estimates of genetic load in humans suggest each individual carries 250 to 700 heterozygous loss-of-function variants, and population genetic analyses using whole-genome sequencing have confirmed that purifying selection removes only a fraction of mildly deleterious mutations each generation, allowing substantial load to persist in large populations.
Genetic load is a central concept in population genetics that quantifies the reduction in mean fitness of a population relative to the fitness of the most fit genotype. The idea was first articulated by J. B. S. Haldane in 1937, who recognised that every deleterious allele maintained in a population imposes a fitness cost that can be expressed as a fraction of the maximum possible population fitness.1 Hermann J. Muller subsequently expanded the concept in his influential 1950 paper "Our load of mutations," arguing that the cumulative burden of deleterious mutations represents a fundamental constraint on population viability and an important selective pressure favouring sexual reproduction and recombination.2 Genetic load is typically expressed as L = 1 − (w̄ / wmax), where w̄ is the mean fitness of the population and wmax is the fitness of the optimal genotype. A population with zero load would consist entirely of individuals with the highest possible fitness, a condition never realised in nature.4, 5
{kind=link}
Mutation load
Mutation load is the component of genetic load attributable to the continuous introduction of deleterious mutations by the imperfect process of DNA replication. At mutation-selection balance, where the rate at which new deleterious alleles enter the population equals the rate at which they are removed by natural selection, the mean fitness reduction caused by mutation is approximately equal to the genomic deleterious mutation rate U, regardless of the fitness effect of individual mutations. This result, known as the Haldane-Muller principle, means that a species with a high per-genome mutation rate carries a proportionally high mutation load even if most individual mutations are only mildly deleterious.1, 2, 5
The genomic deleterious mutation rate has been estimated for a range of organisms. In Drosophila melanogaster, Mukai and colleagues estimated approximately 0.5 to 1.0 deleterious mutations per diploid genome per generation using mutation-accumulation experiments, a finding that suggested substantial standing mutation load.14 In humans, estimates of the deleterious mutation rate range from approximately 1.0 to 3.0 per diploid genome per generation, depending on the fraction of the genome assumed to be functionally constrained and the threshold for defining deleteriousness.9, 15 These estimates imply that each human generation introduces a substantial number of new mildly harmful alleles, most of which are eliminated by purifying selection over subsequent generations but not before contributing to the standing genetic load of the population.10
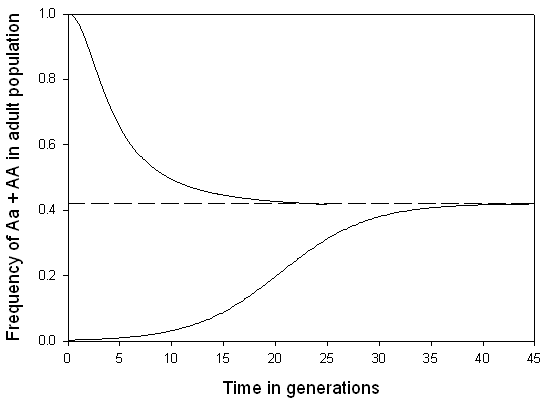
Segregation load
Segregation load arises when heterozygous genotypes are maintained in a population by balancing selection, such as heterozygote advantage (overdominance), but Mendelian segregation continuously regenerates less-fit homozygotes. The classic example is the sickle-cell haemoglobin polymorphism in human populations where malaria is endemic: heterozygotes for the HbS allele have increased resistance to Plasmodium falciparum malaria relative to HbA homozygotes, but HbS homozygotes suffer severe sickle-cell disease. The population maintains both alleles because the heterozygote has the highest fitness, yet every generation a predictable fraction of offspring are HbS homozygotes who pay the fitness cost of the disease. This cost is the segregation load.4, 5
More generally, segregation load exists wherever alleles are maintained by heterozygote advantage at one or more loci. Crow and Kimura showed that if overdominance operates at many loci simultaneously, the segregation load can become very large, because the probability of any individual being heterozygous at all relevant loci decreases exponentially with the number of loci. This argument was central to the "classical" versus "balance" debate in population genetics during the 1960s: the classical school argued that most loci are fixed for a single allele (minimising segregation load), while the balance school maintained that extensive polymorphism is maintained by balancing selection.5 The resolution came partly from the recognition that most molecular polymorphism is selectively neutral or nearly so, as proposed by Motoo Kimura's neutral theory, which implied that the high levels of genetic variation observed at the molecular level do not necessarily generate correspondingly high segregation load.5, 6
Drift load and substitutional load
Drift load is the component of genetic load caused by genetic drift fixing deleterious alleles or losing beneficial alleles in finite populations. In small populations, the stochastic fluctuations of allele frequencies can overpower the deterministic force of selection, allowing mildly deleterious mutations to drift to fixation. Kimura, Maruyama, and Crow demonstrated that the rate of fixation of deleterious mutations increases as effective population size decreases, so that small populations accumulate a disproportionately large drift load.6 This effect is compounded in asexual organisms by Muller's ratchet, the irreversible accumulation of deleterious mutations in the absence of recombination, which provides a potential explanation for the long-term advantage of sexual reproduction.8
Substitutional load (also called the cost of natural selection) was described by Haldane in 1957 as the cumulative fitness cost a population pays when it replaces one allele with another through directional selection.3 Haldane calculated that each gene substitution requires a certain number of "selective deaths" — individuals that fail to reproduce because they carry the old, less-fit allele — and that this cost limits the number of loci that can undergo simultaneous adaptive substitution. He estimated that a typical vertebrate population could sustain on the order of one gene substitution every 300 generations, implying that rapid adaptive evolution at many loci simultaneously would impose an unsustainable fitness cost. This "Haldane's dilemma" was influential in motivating Kimura's neutral theory, which proposed that most molecular evolution involves selectively neutral substitutions that do not incur a fitness cost.3, 5
Empirical estimates in humans and model organisms
Whole-genome sequencing has made it possible to directly estimate the burden of deleterious variants carried by individuals and populations. Data from the 1000 Genomes Project revealed that a typical human genome contains approximately 250 to 700 heterozygous loss-of-function variants affecting protein-coding genes, including roughly 20 genes that are completely inactivated (knocked out) in each individual.16, 17 Most of these loss-of-function variants are rare, consistent with purifying selection acting to keep them at low frequency, but their sheer number across the genome means that every individual carries a non-trivial genetic load.16
Comparative genomic approaches have estimated the distribution of fitness effects (DFE) of new mutations by analysing patterns of divergence between species and polymorphism within species. Eyre-Walker and Keightley estimated that roughly 38% of amino acid-changing mutations in hominids are effectively neutral, approximately 27% are mildly deleterious, and approximately 35% are strongly deleterious and rapidly eliminated by selection.9, 10 The mildly deleterious class is of particular importance for genetic load because these mutations persist in the population long enough to contribute to standing variation but are too harmful to be truly neutral. Genomic constraint scores such as GERP++ have confirmed that a substantial fraction of the human genome — approximately 4 to 5% — is under measurable purifying selection, indicating that mutations at these sites are likely to be deleterious.18
Significance and ongoing debates
The concept of genetic load has influenced several major theoretical debates in evolutionary biology. The paradox of high mutation load in organisms with large genomes was one motivation for Kondrashov's hypothesis that sexual reproduction evolved in part to purge deleterious mutations more efficiently through synergistic epistasis, wherein the combined fitness effect of multiple deleterious mutations is greater than the sum of their individual effects. Under such epistasis, recombination can bring together deleterious alleles into the same genome, creating low-fitness individuals that are efficiently removed by selection, thereby reducing the equilibrium load.7, 8
Genetic load also has practical implications for conservation biology. Populations that have experienced severe bottlenecks or sustained small population sizes are expected to accumulate drift load, potentially reducing their long-term viability. Empirical studies of inbred livestock populations and endangered species have documented fitness declines attributable to the accumulation of homozygous deleterious alleles, a phenomenon termed inbreeding depression that represents a specific manifestation of genetic load in small populations.6, 12 Modern genomic tools now allow researchers to estimate the deleterious mutation burden in threatened species and to assess whether genetic rescue — the introduction of migrants from genetically divergent populations — can reduce load by restoring heterozygosity and masking recessive deleterious alleles.16
The precise magnitude of genetic load in humans remains debated, in part because it depends on assumptions about the fraction of the genome that is functional, the shape of the distribution of fitness effects, and the extent of epistatic interactions among deleterious alleles. Genome-wide association studies have identified thousands of common variants with small effects on complex traits, raising questions about whether these variants represent a form of standing genetic load or are maintained by balancing selection or pleiotropy.10, 13 As whole-genome sequencing becomes routine and the functional consequences of individual variants are better characterised, the empirical measurement of genetic load will continue to refine one of population genetics' most fundamental theoretical constructs.11, 16
References
A spectrum of free fitness effects estimated from a pairwise comparison of human-chimpanzee genes
A general multi-trait approach for estimating the proportion of new mutations that are deleterious
Identifying a high fraction of the human genome to be under selective constraint using GERP++