Overview
- Mitochondrial DNA and Y-chromosome analysis trace all living humans to ancestral populations in Africa, with a major dispersal out of Africa occurring roughly 60,000–70,000 years ago that seeded every inhabited continent.
- Ancient DNA recovered from fossil specimens has transformed understanding of prehistoric migration, revealing multiple waves of movement, substantial admixture between anatomically modern humans and archaic populations such as Neanderthals and Denisovans, and previously unknown ghost lineages.
- Large-scale genomic projects including the 1000 Genomes Project and the Human Genome Diversity Project have catalogued the fine-grained population structure of our species, demonstrating that genetic diversity is greatest in Africa and declines systematically with geographic distance from the continent, consistent with serial founder effects.
The human species is, in genetic terms, strikingly young and remarkably uniform. Despite superficial differences in appearance among populations on different continents, Homo sapiens displays less genetic variation than many other primate species, a consequence of its recent shared ancestry and the powerful compressing effect of serial founder events during its global dispersal from Africa.6 Population genetics—the study of allele frequencies, their changes over time, and the processes that shape them—has in the past four decades been transformed from a discipline reliant on a handful of blood-group markers into one capable of interrogating hundreds of millions of genomic positions simultaneously. That transformation has made it possible to reconstruct human migration history with an unprecedented combination of geographic resolution and temporal depth, writing a detailed molecular chronicle of how our species colonised every habitable corner of the Earth.5, 6
Mitochondrial Eve and Y-chromosomal Adam
Two components of the human genome are passed down without recombination and therefore retain an unbroken genealogical signal across generations: mitochondrial DNA (mtDNA), inherited exclusively through the maternal line, and the non-recombining portion of the Y chromosome (NRY), inherited exclusively through the paternal line. Because they do not shuffle genetic material with each generation, mtDNA and NRY sequences accumulate mutations at roughly clock-like rates, allowing researchers to construct genealogical trees whose branch lengths correspond to elapsed time. Every branch of every such tree eventually coalesces at a single ancestral sequence—the "mitochondrial Eve" or "Y-chromosomal Adam"—the most recent common matrilineal or patrilineal ancestor, respectively, of all living humans.2
The concept of mitochondrial Eve, introduced in a 1987 paper by Cann, Stoneking, and Wilson and refined in subsequent studies including Vigilant and colleagues' 1991 analysis, does not imply that this individual was the only woman alive at the time; it means only that all other contemporary maternal lineages have subsequently gone extinct through chance failure to leave female descendants.2 Analyses of mitochondrial DNA place the coalescence of all living human mtDNA lineages in Africa, with divergence times estimated between roughly 100,000 and 200,000 years ago, consistent with the known antiquity of anatomically modern humans in the fossil record.2 Crucially, the deepest-branching mtDNA haplogroups—designated L0 and L1—are found exclusively in Africa, a pattern that places the root of the human mitochondrial tree unambiguously on that continent.22
Y-chromosomal phylogenies are constructed from thousands of single-nucleotide polymorphisms (SNPs) on the NRY and provide a parallel paternal record. A comprehensive 2001 study by Ke and colleagues analyzed the Y chromosomes of over 12,000 men across East Asia and found that every Y chromosome belonged to haplogroups ultimately derived from African lineages, providing compelling evidence against the hypothesis of regional continuity from archaic populations in Asia.1 The deepest-branching Y-chromosome haplogroup, haplogroup A, is found at highest frequencies among the San people of southern Africa and the Sandawe and Hadza of East Africa—populations whose extraordinary linguistic and cultural antiquity is now matched by genetic evidence of their deep genealogical roots.22
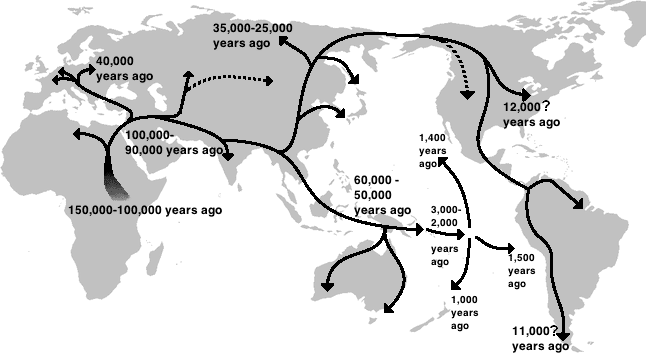
The out-of-Africa dispersal
{kind=link}
Although Homo sapiens first appeared in Africa at least 300,000 years ago, the ancestors of all non-African populations descend from a relatively small group that left the continent during the Late Pleistocene. Genetic evidence consistently places the primary dispersal event at roughly 60,000 to 70,000 years ago, a timing that aligns with the rapid spread of the Later Stone Age toolkit across South Asia, Southeast Asia, and eventually Australia.19, 6 The magnitude of the population bottleneck associated with this dispersal is reflected in the comparatively low genetic diversity of non-African populations: by essentially every metric—heterozygosity, haplotype diversity, number of alleles per locus—non-Africans are a genetically impoverished subset of the variation present in Africa.6
The route or routes of the initial dispersal have been debated extensively. The southern coastal route hypothesis, synthesized by Macaulay and colleagues from mtDNA haplogroup distributions in 2005, proposes that a single founding population followed a beachcombing strategy along the Indian Ocean coastline, moving from the Horn of Africa across the Arabian Peninsula, through South Asia, and eventually into Southeast Asia and Australasia—all within a geologically brief period of perhaps 10,000 to 15,000 years.19 Under this model, the extraordinary speed of dispersal was enabled by exploitation of coastal and estuarine resources that provided a reliable food supply without requiring adaptation to unfamiliar inland environments. An alternative northern route, crossing the Sinai Peninsula and following river valleys through the Levant and into Central Asia, has also been proposed, and some researchers argue for multiple dispersals using both corridors at different times.6
The discovery of a 45,000-year-old modern human genome from Ust'-Ishim in western Siberia, reported by Fu and colleagues in 2014, provided crucial temporal calibration for the dispersal chronology.8 The Ust'-Ishim individual's genome showed long Neanderthal-derived haplotype segments, indicating that admixture with Neanderthals had occurred not long before this individual lived—consistent with interbreeding in the Middle East or Central Asia as dispersing modern humans first encountered Neanderthal populations. By measuring the length distribution of these Neanderthal-derived segments (which shorten with each generation of recombination), Fu and colleagues estimated that the admixture event occurred 50,000 to 60,000 years before the Ust'-Ishim individual's death, placing the initial out-of-Africa movement squarely in the 60,000–70,000-year window suggested by other lines of evidence.8
Admixture with archaic humans
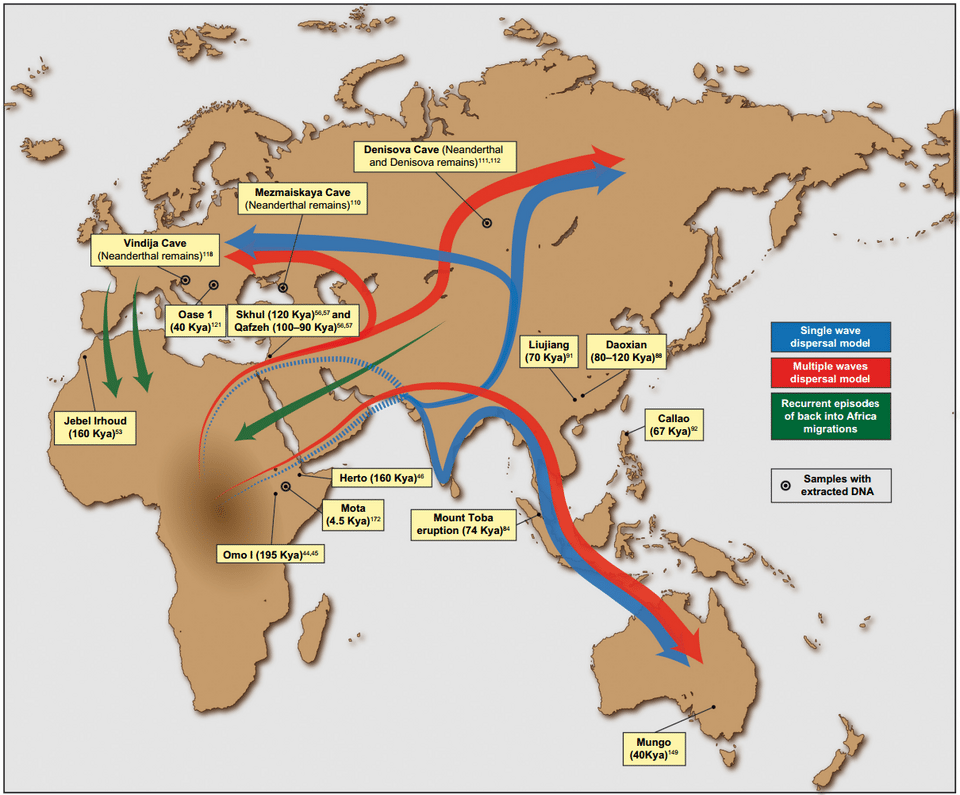
One of the most consequential discoveries of the ancient DNA era is the demonstration that dispersing modern humans did not simply replace archaic populations they encountered but interbred with them, incorporating archaic genomic material that persists in the genomes of their living descendants. The first unambiguous evidence came in 2010 from Green and colleagues' draft sequence of the Neanderthal genome, assembled from bone fragments from Vindija Cave in Croatia.3 Comparing the Neanderthal genome to those of five modern humans from geographically diverse populations, Green and colleagues found that individuals of non-African ancestry share slightly more alleles with Neanderthals than do Africans—a pattern most parsimoniously explained by gene flow from Neanderthals into the ancestors of non-Africans after those ancestors left Africa but before they dispersed across Eurasia.3 Subsequent analyses using high-coverage Neanderthal genomes have refined the estimate of Neanderthal ancestry in non-African populations to approximately 1 to 4%.11
{kind=link}
A second archaic population, the Denisovans, was identified from a single finger bone and two molar teeth from Denisova Cave in the Altai Mountains of Russia. DNA from the finger bone, sequenced in 2010 by Reich and colleagues, revealed a population that was genetically distinct from both modern humans and Neanderthals, representing a sister lineage to the Neanderthals that had diverged from them several hundred thousand years ago.4 Although Denisovan fossils have been found only at Denisova Cave and a single jaw from the Tibetan Plateau, their genetic legacy is widespread: populations in Melanesia, Aboriginal Australians, and some Southeast Asian groups carry 4 to 6% Denisovan-derived DNA, while populations elsewhere in Asia and the Americas carry smaller but detectable fractions.4, 12
The functional significance of archaic introgression has been demonstrated in several cases. Huerta-Sánchez and colleagues showed in 2014 that the high-altitude adaptation of Tibetan populations—specifically a variant of the gene EPAS1 (encoding hypoxia-inducible factor 2-alpha) that allows efficient oxygen transport at elevation—was acquired from Denisovans through introgression rather than evolving de novo in the Tibetan lineage.17 The Tibetan EPAS1 haplotype is nearly identical to the corresponding sequence in the Denisovan genome, strongly supporting an introgressive origin. This finding illustrates how archaic admixture can provide modern humans with adaptive variants that had already been shaped by selection in populations long adapted to local environments—a form of genetic inheritance that may have accelerated adaptation in the populations that received it.17
The peopling of Sahul and Australia
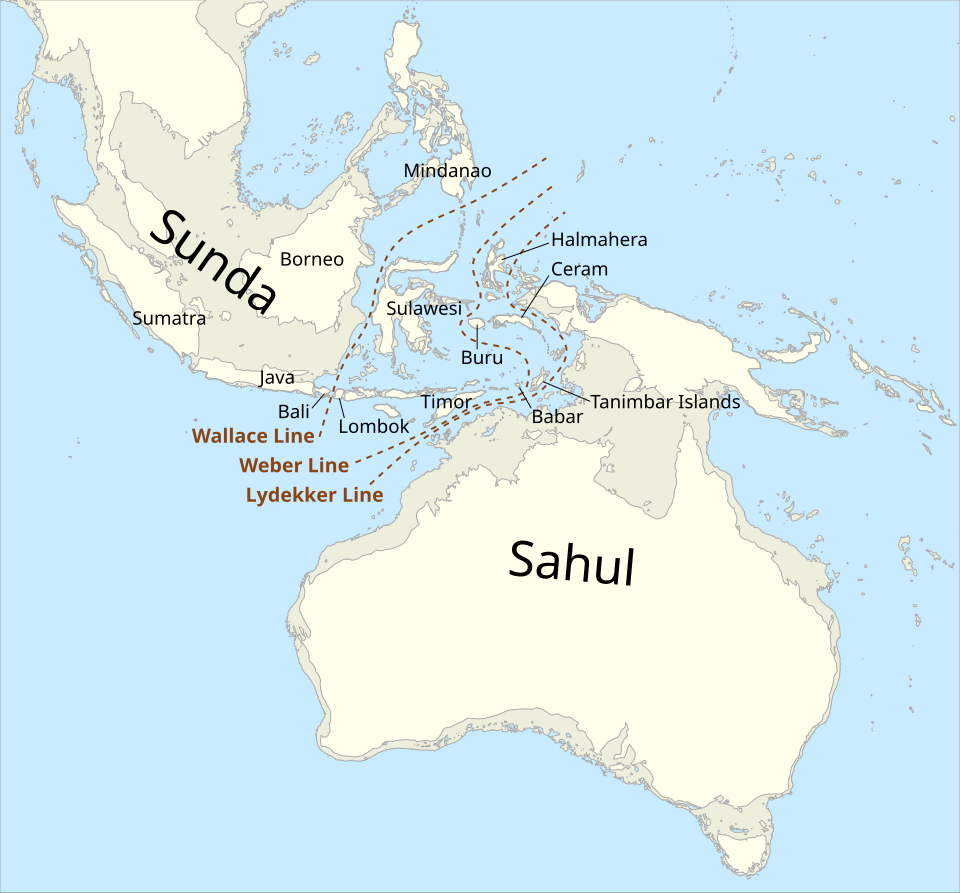
The colonization of Sahul—the landmass comprising present-day Australia, New Guinea, and Tasmania, which were connected during glacial sea-level low-stands—represents one of the most remarkable feats of prehistoric navigation, requiring a maritime crossing of at least 70 to 90 kilometres even when sea levels were at their lowest.12 Archaeological evidence, including charcoal from hearths at Lake Mungo in New South Wales and shell middens in northern Australia, places modern humans in Australia by at least 47,000 to 50,000 years ago, with some sites potentially indicating an even earlier presence approaching 65,000 years ago.12
{kind=link}
Genetic evidence corroborates a very early arrival. The genomes of Aboriginal Australians and Papuans are distinguished by their high proportion of Denisovan ancestry, which is essentially absent from mainland Asian populations, suggesting that the ancestors of Australasians diverged from other non-African populations before the latter dispersed across Eurasia, thereby avoiding further dilution of Denisovan ancestry by later gene flows.4, 12 Genomic analyses by Skoglund and colleagues in 2016 demonstrated that Papuan and Aboriginal Australian populations carry a genomic signature of a distinct ancestry that appears to have split from other non-Africans shortly after the out-of-Africa dispersal, consistent with an early and rapid colonization of Sahul by a founding population that remained relatively isolated for tens of thousands of years.12
The ancient DNA record of Southeast Asia and the Pacific has been substantially enriched by analyses of skeletal remains from island Southeast Asia, reported by McColl and colleagues in 2018. These data reveal that the pre-Neolithic inhabitants of the region were related to present-day Aboriginal Australians and Papuans—a "deep Asian" ancestry—and were largely replaced or admixed with incoming populations associated with the spread of agriculture from mainland East Asia beginning around 4,000 years ago.13 The complex layering of ancestries in island Southeast Asia thus records at least two major waves of migration: an initial colonization by groups ancestral to present-day Australo-Papuans, followed millennia later by the Austronesian expansion.13
The Austronesian expansion and Lapita culture
The Austronesian-speaking peoples represent the most geographically extensive language family in the world, encompassing over 1,200 languages spread from Madagascar in the west to Easter Island (Rapa Nui) in the east and from Taiwan in the north to New Zealand in the south. Linguistic, archaeological, and genetic evidence converge on Taiwan as the homeland from which Austronesian-speaking populations began expanding southward approximately 4,000 to 5,000 years ago, reaching the Philippines by around 3,500 years ago, the western Pacific by 3,200 years ago, and the far reaches of Polynesia between approximately 700 and 1,000 years ago.20
The archaeological signature of the initial Austronesian expansion into the Pacific is the Lapita cultural complex, characterised by a distinctive style of dentate-stamped pottery first appearing around 3,500 to 3,200 years ago in the Bismarck Archipelago of Papua New Guinea and subsequently spreading rapidly through the Solomon Islands, Vanuatu, New Caledonia, Fiji, Tonga, and Samoa.21 The Lapita people were accomplished open-ocean navigators who used outrigger canoes and sophisticated wayfinding knowledge of stars, currents, and birds to traverse vast expanses of the Pacific. Kirch's analysis of Lapita cultural distribution identified the complex as a horizon marker for the initial colonization of Remote Oceania—the islands beyond the range of earlier Papuan maritime expansion.21
Genomic analyses have clarified the ancestry of Lapita-associated individuals. Ancient DNA from Vanuatu and Tonga, reported by Skoglund and colleagues in 2016, showed that the earliest Lapita individuals carried almost entirely East Asian-related ancestry with negligible Papuan-related ancestry, contradicting earlier models that envisioned substantial population mixing with resident Papuan populations before the expansion into Remote Oceania.12 The present-day Polynesian populations—descended from Lapita communities in western Polynesia—carry a mixture of this founding East Asian ancestry and Papuan ancestry acquired through later contact with Melanesian populations, a blending that occurred after the initial Lapita diaspora but before the final settlement of eastern Polynesia.12
Approximate timing of major human dispersal events6, 19, 7, 20, 24
| Event | Approximate date | Primary evidence |
|---|---|---|
| Out-of-Africa dispersal | 60,000–70,000 years ago | mtDNA haplogroups, ancient DNA haplotype length |
| Colonization of Sahul (Australia/New Guinea) | 47,000–65,000 years ago | Archaeology (Lake Mungo), Denisovan ancestry fraction |
| Entry into the Americas (Beringia) | ~20,000–25,000 years ago (standstill) | mtDNA haplogroups A–D, X; ancient DNA |
| Rapid peopling of the Americas south of ice sheets | 15,000–16,000 years ago | Monte Verde, Paisley Caves, Clovis sites |
| Lapita expansion / initial Remote Oceania colonization | 3,200–3,500 years ago | Lapita pottery, ancient DNA, linguistic phylogenies |
| Settlement of eastern Polynesia and Hawaii | 700–1,000 years ago | Radiocarbon dating, oral tradition, genomics |
The peopling of the Americas
The colonization of the Americas was the last major continental-scale dispersal in human prehistory and has been the subject of intense archaeological and genetic debate. The prevailing model, supported by convergent lines of genetic evidence, posits that the ancestors of all Native American populations passed through Beringia—the landmass connecting Siberia to Alaska during glacial periods of lowered sea level—and that this migration was preceded by a period of isolation in Beringia lasting several thousand years before populations moved into the Americas proper.24
The Beringian standstill hypothesis, formalized genetically by Tamm and colleagues in 2007, interprets the pattern of mitochondrial DNA haplogroup diversity in Native American populations as evidence that the founding population was isolated in Beringia for an extended period—estimated at 5,000 to 15,000 years—before dispersing southward when an ice-free corridor between the Laurentide and Cordilleran ice sheets opened, or alternatively along a coastal route, approximately 15,000 to 16,000 years ago.24 Native American mtDNA belongs to five founding haplogroups—A, B, C, D, and X—all of which are ultimately derived from Asian maternal lineages, consistent with a northeastern Asian origin for the ancestral population.24
Archaeological evidence for pre-Clovis human presence south of the ice sheets has substantially strengthened the case for an early coastal entry route that predates the opening of an inland ice-free corridor. The site of Monte Verde in southern Chile, excavated by Tom Dillehay and his team, contains cultural and organic materials radiocarbon-dated to approximately 14,500 to 14,800 years ago—making it several centuries older than the earliest securely dated Clovis sites in North America.10 Because Monte Verde is located at the southern tip of South America, populations must have been present in the Americas well before this date to have traversed the continent from north to south, implying an initial entry by at least 16,000 years ago and possibly earlier.10 Molecular human coprolites from the Paisley Caves in Oregon, genetically attributed to haplogroup B2 Native Americans by Gilbert and colleagues in 2008, provide additional pre-Clovis evidence dated to approximately 14,300 years ago in North America.9
The genetic evidence for the peopling of the Americas also reveals complexity that a single-wave model cannot accommodate. Raghavan and colleagues' 2015 analysis of ancient and modern Native American genomes identified a signal of differential ancestry that distinguishes Amazonian populations and certain other South American groups from most other Native Americans—a "Population Y" signature related to Australo-Papuans and Andamanese rather than to Northeast Asians.7 Whether this represents a distinct founding wave that entered the Americas separately from the main ancestral population, or reflects gene flow from a source population with mixed East Asian and "deep Asian" ancestry, remains under active investigation. Subsequent to the initial colonization, Arctic populations including the Paleo-Eskimos and later Neo-Eskimos represent additional migrations from Asia into the Americas, documented by ancient DNA from sites across the Arctic.25
The Bantu expansion in Africa
Within Africa, the most geographically extensive population movement of the Holocene was the Bantu expansion, which spread Bantu-speaking agriculturalists from a homeland in the Benue-Cross River region of present-day Cameroon and Nigeria across sub-Saharan Africa beginning approximately 4,000 to 5,000 years ago. The expansion proceeded in two broad streams: a western stream moving along the equatorial coast into the Congo Basin, and an eastern stream skirting the East African Rift Valley before sweeping southward through eastern and southern Africa to reach the tip of the continent approximately 2,000 years ago.22
The Bantu expansion represents a dramatic replacement and admixture event in African population history, comparable in continental scale to the Neolithic expansion of farming populations into Europe from Anatolia. Tishkoff and colleagues' landmark 2009 study of African genetic structure, based on 1,327 polymorphic markers genotyped in over 3,000 Africans from 113 populations, documented the extent to which Bantu-speaking populations now dominate the genetic landscape of sub-Saharan Africa outside of relict hunter-gatherer groups such as the Khoisan, Hadza, and Mbuti Pygmies.22 The Khoisan populations of southern Africa, despite their geographic isolation from the postulated origin point of Bantu expansion, carry the deepest-branching mitochondrial and autosomal lineages of any living humans, serving as a genomic window into the structure of African populations before the agricultural diaspora.22
Founder effects and genetic drift
A fundamental insight of population genetics, formalised in the "serial founder effect" model of human dispersal, is that each successive colonization event along the path of human migration involved only a subset of the genetic diversity present in the source population. The founders of a new colony are, by definition, a small and non-representative sample of the parent population; if they do not subsequently receive immigrants from elsewhere, their descendants will carry only the genetic variants that happened to be present in that small founding group.6 Applied iteratively along a chain of colonizations from Africa through the Middle East, Central Asia, and beyond, this process predicts that genetic diversity should decrease in a linear fashion with geographic distance from Africa—a prediction that has been confirmed in every major genomic survey of human populations.6
Li and colleagues' 2008 analysis of over 650,000 SNPs in 938 individuals from 51 worldwide populations, using data from the Human Genome Diversity Project panel, provided the most comprehensive demonstration of this pattern to that date.6 Heterozygosity—the probability that two alleles drawn at random from a population differ from each other, a standard measure of genetic diversity—declined with distance from Africa measured along plausible overland migration routes. Sub-Saharan African populations showed the highest heterozygosity, followed in decreasing order by Middle Eastern, Central Asian, European, East Asian, American, and Oceanian populations. This gradient is not explained by climate, diet, or any variable other than the cumulative compounding of founder effects along the dispersal pathway.6
Approximate heterozygosity by region, declining with distance from Africa6
Genetic drift—the random fluctuation of allele frequencies in finite populations—acts most powerfully in small populations and over many generations compounds into dramatic differentiation between isolated groups. Some of the most striking examples of extreme drift are found in island populations and religious isolates, where alleles that were rare or absent in the founding population have risen to high frequency, and where certain alleles common in the source population have been entirely lost. The Amish of North America and the Ashkenazi Jewish population of Eastern Europe both provide medically important examples: each community was founded from a relatively small group and has practiced significant endogamy, resulting in elevated frequencies of specific recessive disease alleles that were either present at low frequency in the founders or arose by mutation after the founding.15
Adaptation signatures in human genomes
Migration into novel environments imposed new selective pressures on dispersing populations, leaving detectable signatures in the genome in the form of haplotype blocks that have risen to high frequency faster than drift alone can explain. The study of these signatures—through methods including extended haplotype homozygosity (EHH) tests, population differentiation statistics such as FST, and composite likelihood ratio tests—has identified dozens of loci subject to strong positive selection in specific populations.18
Among the best-characterized examples is the evolution of lactase persistence—the retention of high lactase enzyme activity in adults, enabling the digestion of lactose in raw milk. In populations without a history of cattle pastoralism, the LCT gene encoding lactase is down-regulated after weaning, as it is in all other mammals. The derived allele conferring adult lactase persistence in European populations (the −13910*T variant) rose to frequencies exceeding 70 to 80% in northern European populations within approximately 5,000 to 10,000 years following the adoption of cattle husbandry, representing one of the strongest signals of recent positive selection in the human genome.16 Tishkoff and colleagues demonstrated in 2007 that different mutations in the same regulatory region confer lactase persistence in East African pastoralist populations—the Oromo and Beja of Ethiopia and the Maasai and Sukuma of Tanzania—constituting a striking case of convergent evolution driven by the independent cultural adoption of dairying.18
Skin pigmentation provides another instructive case study. Populations residing at low latitudes, where UV radiation is intense, generally have darker skin pigmentation produced by high concentrations of eumelanin, which protects against UV-induced DNA damage and folate photolysis. Populations at high latitudes, where UV radiation is lower and the biosynthesis of vitamin D from sunlight is limiting, have generally evolved lighter pigmentation that facilitates vitamin D production. The genetic architecture of pigmentation differences among populations involves scores of loci, including SLC24A5, SLC45A2, HERC2/OCA2, and MC1R, and analysis of ancient DNA has shown that the light-skinned alleles common in modern Europeans were largely absent from Mesolithic European hunter-gatherers, only reaching high frequency with the Neolithic and Bronze Age expansions of farmers from Anatolia and steppe herders from the Pontic-Caspian region.5
Altitude adaptation in Tibetans, mentioned above in the context of Denisovan introgression, exemplifies how archaic admixture and local selection can interact. The EPAS1 variant acquired from Denisovans allows carriers to maintain haemoglobin concentrations closer to sea-level norms at high altitude, avoiding the polycythaemia (excess red blood cell production) that is associated with altitude sickness and cardiovascular complications in other populations that have moved to high elevation.17 This variant has been driven to very high frequency in Tibetans by strong positive selection, yet its Denisovan origin illustrates that the raw material for adaptation was not a de novo mutation in the Tibetan lineage but a variant that had been present in the Denisovan lineage, presumably under selection for high-altitude survival in populations inhabiting the Tibetan Plateau before the arrival of modern humans.17
The ancient DNA revolution
The field of ancient DNA (aDNA) was transformed beginning around 2010 by the development of high-throughput next-generation sequencing methods capable of recovering and assembling highly degraded, short DNA fragments from fossil specimens. Prior to this methodological revolution, aDNA studies were largely limited to short segments of mitochondrial DNA retrieved from relatively recent specimens in cold, dry environments. The new methods made it possible to sequence complete or near-complete genomes from specimens tens of thousands of years old, dramatically expanding both the temporal depth and the geographic breadth of the fossil genomic record.3, 4
Ancient DNA analyses have fundamentally revised understandings of population history that could not be inferred from modern genomes alone. In Europe, aDNA studies have documented at least three major episodes of population replacement or large-scale admixture during the Holocene: the spread of early Neolithic farmers from Anatolia beginning approximately 9,000 years ago, who largely replaced the resident Mesolithic hunter-gatherers; the subsequent expansion of Yamnaya-related steppe herders from the Pontic-Caspian grasslands around 5,000 years ago, who introduced steppe ancestry into both Europe and South Asia on a massive scale; and continuing gene flows from the Near East and elsewhere that shaped the population structure of historical-period European societies.5 These episodes were invisible in modern European genomes, which represent the blended outcome of all these historical processes, but are legible in the genomes of ancient individuals sampled from before, during, and after each event.5
The ancient genomic record of the Americas, reconstructed from specimens spanning from the late Pleistocene through the historical period, has similarly revealed complexity invisible in modern populations. Rasmussen and colleagues' 2010 sequencing of a 4,000-year-old Saqqaq Paleo-Eskimo from Greenland showed this individual to belong to a migration wave distinct from both Na-Dene-speaking populations and the main founding population of the Americas, consistent with a separate and later migration of Arctic-adapted peoples from northeastern Asia.25 These findings, combined with the broader ancient DNA record, indicate that the Americas were not colonized by a single founding event but by a series of migrations that occupied different ecological niches and left traces in the genomic structure of present-day Indigenous populations.7
Large-scale genomic projects
Two large international consortia have done more than any other projects to characterise the global genetic structure of living humans at high resolution. The Human Genome Diversity Project (HGDP), initiated in the 1990s, systematically collected DNA from individuals representing over 50 populations from around the world, providing a globally representative reference panel that has been used in hundreds of studies of human population history, selection, and disease genetics.6 The HGDP panel was used by Li and colleagues to produce the comprehensive analysis of heterozygosity gradients described above, and it remains a foundational resource for the field.6
The 1000 Genomes Project, completed in 2015 and reported in a landmark Nature paper by the project consortium, went substantially further by sequencing the whole genomes of 2,504 individuals from 26 populations at sufficient depth to characterise not only common variants but also low-frequency and rare variation.5 The project identified approximately 88 million SNPs, 3.6 million short insertions and deletions, and 60,000 structural variants—the most comprehensive catalogue of human genetic variation assembled to that point. Population-specific analyses within the 1000 Genomes framework confirmed the serial founder effect gradient of diversity, documented the extent of linkage disequilibrium (the correlation of allele frequencies at nearby sites due to shared genealogical history) across populations, and provided a reference panel now essential for imputing variants in genome-wide association studies of complex disease.5
These datasets have also refined understanding of population structure within continental regions. Within Africa, Tishkoff and colleagues identified 14 ancestral population clusters whose geographic distributions largely corresponded to linguistic groupings, reflecting the deep correspondence between cultural and biological descent that characterises much of African prehistory.22 Within the Americas, Y-chromosome analyses by Bianchi and colleagues documented distinct haplogroup distributions between North and South American populations that preserve the demographic signal of separate colonization events and post-colonization isolation, while mtDNA and genomic data from Raghavan and colleagues have continued to elaborate the fine-scale structure of Native American population history.23, 7
Taken together, the convergent evidence from maternal lineages, paternal lineages, autosomes, and ancient DNA paints a consistent portrait of a species with deep African roots, a relatively recent global dispersal, and a history of migration, admixture, isolation, and adaptation that has left its signature in every genome on Earth. The study of human population genetics is not merely an exercise in reconstructing the past; it is the foundation for understanding the present distribution of genetic disease risk, drug response variation, and the evolutionary forces that continue to shape human biology in real time.5, 6
References
Pre-Clovis human occupation of the Americas identified by human coprolites at the Paisley Caves
The expansion of farming peoples and the role of the Neolithic demographic transition in Y-chromosome evolution in Europe
Lactase persistence alleles reveal partial convergent evolution and differing selective pressures in humans